Autor: Paulo Cesar Naoum |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Fenótipos, genótipos e haplótipos A doença causada pelas células falciformes se caracteriza por um conjunto de sinais e sintomas provocados pela deformação de milhões de eritrócitos com hemoglobina S (Hb S) induzidos pela desoxigenação. Esses eritrócitos com Hb S quando oxigenados (oxi-Hb S) se apresentam morfologicamente normais ou discóides no sangue; eventualmente podem ser observados alguns eritrócitos afoiçados ou falcizados (figura 6.1). Entretanto, há situações em que o processo de falcização é muito intenso devido à elevada concentração de Hb S desoxigenada (desoxi-Hb S), conforme mostra a figura 6.2. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
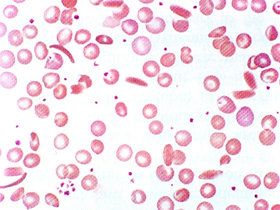 Figura 6.1 – Esfregaço de sangue periférico de paciente com anemia falciforme controlada, mostrando maior número de eritrócitos discóides em relação aos falcizados. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
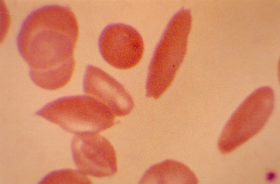
Figura 6.2 – Esfregaço de sangue periférico de paciente com anemia falciforme, coletado durante crise dolorosa, mostrando eritrócitos discóides com equinocitose e vários eritrócitos falcizados. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
O primeiro relato a respeito da doença das células falciformes data de 1910, quando J.B. Herrick publicou um artigo científico na revista americana Arquivo de Medicina Interna sobre a presença de eritrócitos com a forma de foice em um paciente negro com anemia grave, icterícia, e fortes dores nas articulações. A partir desse artigo outras publicações de casos semelhantes foram divulgadas, inclusive identificando a origem hereditária das células falciformes (Neel, 1949). Entretanto, todas essas comunicações destacavam apenas a diversidade clínica da doença com relação à gravidade e ao desenvolvimento da patologia. Todavia, todos os trabalhos descritos até 1949 apresentavam a doença das células falciformes pelas características visíveis do processo anêmico específico, associado à presença de células falcizadas no esfregaço de sangue periférico e com notável prevalência entre negros ou descendentes africanos. Essa associação de características muito semelhantes fornecia o fenótipo de uma doença que era causada pelas células falciformes, e por isso essa patologia hematológica passou a ser conhecida por doença das células falciformes. Não obstante, ainda continuava sem explicação convincente a grande diversidade clínica e hematológica entre os doentes falcêmicos. No ano de 1949 Linus Pauling juntamente com seus colegas de laboratório conseguiram separar por meio de eletroforese a hemoglobina anormal que causava a falcização eritrocitária, que foi denominada por "sickle hemoglobin" (ou hemoglobina falcizante). Para facilitar a divulgação científica desta hemoglobina anormal tomou-se por referência a primeira letra da palavra "sickle", surgindo então a Hb S. O processo eletroforético foi de grande importância para o entendimento clínico, genético e hematológico dos doentes falcêmicos, pois sua aplicação permitiu explicar que a diversidade clínica e hematológica estava relacionada com a concentração da Hb S, bem como sua associação com outras hemoglobinas. Assim foram identificadas várias situações da doença em que a concentração da Hb S era variável. Hb S com concentrações próximas de 95-98% foi geneticamente caracterizada por Hb SS (homozigose da Hb S) e a doença foi especificamente denominada por anemia falciforme. Outros pacientes que, além de células falciformes continham também eritrócitos microcíticos e hipocrômicos, apresentavam em suas eletroforeses a Hb S associada com Hb Fetal, ou Hb F, e por isso identificados como Hb SF, foram caracterizados como doentes da anemia micro-drepanocítica e atualmente são conhecidos por Hb S / talassemia beta ou Hb S / b tal. Com o passar do tempo, o aperfeiçoamento técnico da eletroforese permitiu a identificação de associações de Hb S com outras hemoglobinas variantes, e assim foram descritas as Hb SC, Hb SD e Hb SO Arábia; essas associações foram nominadas por doenças falciformes SC, SD e SO Arábia, respectivamente. Outras interações mais raras como são os casos da anemia falciforme associada à talassemia alfa pela presença de Hb S e Hb H conjuntamente, e entre Hb S e Hb Fetal elevada – caracterizada por persistência hereditária de Hb Fetal (PHHF), foram denominadas por Hb SS / Talassemia alfa e Hb S / PHHF. Finalmente, o caso mais comum é a presença concomitante da Hb S e Hb A (Hb AS) no portador do traço falcêmico, ou heterozigoto falcêmico, clinicamente assintomático e com eritrograma normal, onde a concentração da Hb S é menor que a Hb A. Essas associações de Hb S com outras frações normais e anormais de hemoglobinas, inicialmente estabelecidas por eletroforeses de hemoglobinas, e confirmadas por estudos nos familiares desses pacientes caracterizaram os diferentes genótipos da doença das células falciformes (figura 6.3). Dessa forma, o conjunto dos resultados laboratoriais e sintomas clínicos permitiram diferenciar adequadamente os genótipos de Hb S (tabelas 6.1 e 6.2). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
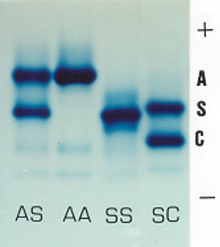
Figura 6.3 – Eletroforese de hemoglobinas em acetato de celulose, pH 8.6, mostrando da esquerda para a direita os genótipos Hb AS (heterozigoto de Hb S); Hb AA (padrão normal); Hb SS (homozigose de Hb S) e Hb SC (dupla heterozigose de Hb S e Hb C). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.1 - Genótipos de Hb S relacionados com alterações laboratoriais. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(*) Principais frações de hemoglobinas
identificadas por eletroforese alcalina. (**) O genótipo da Hb AS não é considerado como doença das células falciformes. Foi incluído apenas para efeito de comparação. O portador de Hb AS é assintomático. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.2 - Alterações eritrocitárias mais comuns nos diferentes genótipos da doença das células falciformes. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(*) Visíveis quando coradas com azul de
crezil brilhante a 1%. (**) Inclui os genótipos S/b 0 Tal. (ou SF) e S/b + Tal. (ou SFA). (***) O genótipo AS não é considerado como doença falciforme. Foi incluído apenas para efeito de comparação. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Durante anos os casos clínicos de doenças das células falciformes foram classificados sem que fossem conhecidas as causas que deram origem à Hb S. Em 1956, Ingram estabeleceu quimicamente pela técnica de "fingerprinting" de peptídeos (figura 6.4) a diferença entre Hb A e Hb S. Constatou que se devia à mutação do sexto aminoácido da globina beta – o ácido glutâmico pela valina, assim representado (Hb S: b 6 Glu ® Val). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
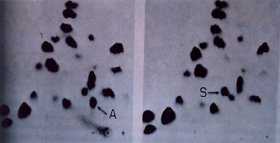
Figura 6.4 – Fingerprints (eletroforese associada a cromatografia em papel de filtro) de péptidos obtidos após digestão de tripsina das globinas de Hb AA e Hb SS. A seta A indica o péptido normal que contém os onze primeiros aminoácidos da globina beta normal. A seta S mostra no digerido tríptico da globina de Hb S que o péptido está em posição diferente à da Hb A (círculo em branco). A análise dos aminoácidos que compõe esse péptido revelou que o sexto aminoácido (ácido glutâmico) fora substituído por outro diferente (valina). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Em 1978 Kan e Dozy descreveram a seqüência de bases nitrogenadas do gene da globina beta normal (Hb A) e da globina beta S (Hb S). Em 1980 esses mesmos pesquisadores estudaram a evolução da Hb S por meio de técnicas de biologia molecular em vários doentes falcêmicos e concluíram que apesar da mutação da Hb S ser a mesma em todos eles, haviam diferenças entre as seqüências de bases nitrogenadas ao longo do agrupamento de genes da globina beta, sem que essas seqüências diversificadas influenciassem na síntese da hemoglobina S. Esses estudos realizados por fracionamento das seqüências de bases induzidos por enzimas extraídas geralmente de bactérias (endonucleases), permitiram estabelecer que a Hb S originou em pelo menos três regiões da África: Senegal, Benin e Bantu. Atualmente acredita-se que além destas três regiões, outras duas regiões, uma na África (Camarões) e outra na Ásia (Arábia e Índia) também sofreram o impacto da mutação da Hb S. Essa diferenciação antropológica identificada por técnicas de biologia molecular entre pessoas doentes com Hb S é conhecida por haplótipos. Até o presente são conhecidos os haplótipos Benin, Bantu, Senegal, Camarões, Árabe-Indiano e Atípicos. Os estudos antropológicos e hematológicos realizados com pacientes com anemia falciforme (Hb SS) e submetidos às análises de seus haplótipos, revelaram que os haplótipos Benin e Bantu estão relacionados com maior grau de anemia quando comparados com os haplótipos Senegal, Camarões e Árabe-Indiano (tabela 6.3), provavelmente devido à persistência da Hb Fetal nesses três haplótipos. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.3 – Relação entre a gravidade da anemia falciforme (Hb SS) com os diferentes haplótipos de Hb S. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(++++) muito grave; (+++) grave; (++) moderado;
(+) discreto |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As células falciformes são mais frágeis que os eritrócitos normais na corrente sangüínea e, por essa razão, vivem menos que o tempo médio de 120 dias. Os eritrócitos falcêmicos tem suas lesões intensificadas na relação direta com o aumento da concentração da Hb S intra-eritrocitária. Assim, na anemia falciforme onde a concentração da Hb S é superior a 90%, o período médio de vida dos eritrócitos falcêmicos é de aproximadamente 30 dias. As causas e conseqüências da falcização, que serão apresentadas com mais detalhes a seguir, se devem de uma forma geral à deformação patológica das células falciformes e à sua precoce destruição motivada pelo ataque da membrana celular por várias espécies de radicais livres gerados durante a degradação da Hb S desoxigenada. Como resposta fisiológica natural à reposição de eritrócitos precocemente retirados da circulação na doença das células falciformes, a eritropoiese medular se torna hiper-ativa. Devido ao desequilíbrio anormal entre a produção de eritrócitos pela medula óssea e a destruição acentuada das células falcêmicas no sangue periférico, ocorre a liberação de células imaturas (reticulócitos) e jovens (eritroblastos) para o sangue circulante, resultando em reticulocitose e presença de eritroblastos no esfregaço de sangue periférico (figuras 6.5 e 6.6). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
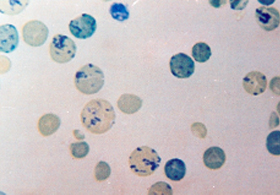
Figura 6.5 – Reticulocitose em amostra de sangue periférico de paciente com anemia falciforme, caracterizando eritropoiese acentuada. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
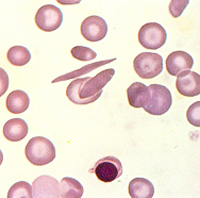
Figura 6.6 – Presença de eritroblasto ortocromático em amostra de sangue periférico de paciente com Hb S/ talassemia beta. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Na corrente sangüínea os eritrócitos falcizam e desfalcizam reversivelmente conforme ganham e liberam o oxigênio. Geralmente há células que, após seguidos processos de falcização e desfalcização, ao se falcizarem, assim permanecem, mesmo que haja oferta de oxigênio; são as células falciformes irreversíveis (figura 6.7). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Figura 6.7 – Célula falciforme irreversível em microscopia eletrônica. A célula falciforme irreversível tem a forma característica de foice, com acentuado alongamento eritrocitário devido à formação de polímeros de moléculas de Hb S desoxigenada. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Quando o número de células falcizadas irreversivelmente permanece abaixo de 5% do conteúdo total de eritrócitos em circulação, o fluxo da circulação de sangue não sofre descontinuidade no seu processo fisiológico. Entretanto, quando o número de células falcizadas irreversivelmente é maior que 5%, tem início o processo de alteração do fluxo sangüíneo. O excessivo número de eritrócitos falcizados irreversivelmente se caracteriza pela sua morfologia achatada e tentacular (figura 6.8) fato que os fazem aderir com maior facilidade ao endotélio vascular, e entre elas próprias, promovendo a obstrução do fluxo sangüíneo especialmente nos pequenos capilares que ligam as veias às artérias. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Figura 6.8 – Microscopia eletrônica de varredura de células falcizadas obtidas de sangue de paciente com anemia falciforme. Observar as formas achatadas e os prolongamentos tentaculares dessas células. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As conseqüências dessas oclusões vasculares são caracterizadas por pequenos infartos teciduais e dores agudas nas regiões atingidas. É evidente que vários fatores têm influência na intensidade das lesões, porém a concentração da deoxi-Hb S intra-eritrocitária se põe como principal indutor desse processo patológico, razão pela qual a diversidade clínica da doença das células falciformes é sempre motivo da necessária qualificação do genótipo da doença. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A
doença falciforme (DF) compreende um grupo de hemoglobinopatias
com repercussão clínica significante, caracterizada
pela herança do gene falciforme em pelo menos um dos pais.
Os principais genótipos que compõem a DF resultam
da homozigose para o gene da Hb S ou anemia falciforme,
e da dupla heterozigose composta entre o gene da Hb S e outras variantes
como Hb C e Hb D (ex.: Hb SC e Hb SD), bem como na interação
com a talassemia beta (ex.: Hb SF ou S/beta talassemia). Além
disso, indivíduos com anemia falciforme podem ser portadores
também de talassemia alfa ou de persistência hereditária
da hemoglobina fetal (PHHF). O conhecimento dos diferentes genótipos
da DF faz-se necessário em virtude de suas implicações
no quadro clínico e prognóstico dos pacientes. O fenótipo
expressado nessa doença também sofre influência
dos diferentes haplótipos do gene falciforme e seus efeitos
pleiotrópicos. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||