Introdução
Autores:pppp Paulo Cesar Naoum Flávio Augusto Naoum |
||||||||||||||||||||||
As
alterações das hemoglobinas envolvem a síntese
estrutural e quantitativa dos aminoácidos que compõem
as diferentes cadeias de globinas, bem como as moléculas
e enzimas que participam da formação do grupo heme. As alterações que ocorrem nas globinas se devem às modificações nos genes responsáveis pelo sequenciamento e estrutura de cada tipo de polipeptídeo de globina, bem como naqueles destinados à regulação quantitativa da síntese equilibrada entre as globinas alfa e beta (b, d e g). Quando um determinado gene apresenta uma de suas bases nitrogenadas substituída por outra diferente, resulta na formação de moléculas de hemoglobinas com características bioquímicas alteradas em relação às hemoglobinas normais e são por isso denominadas hemoglobinas variantes. Por exemplo, a hemoglobina S se deve à introdução do aminoácido valina (Val) no lugar do ácido glutâmico (Glu) na posição número 6 da cadeia polipeptídica da globina beta por meio de um processo conhecido por mutação. Essa anormalidade se deve a uma troca da base nitrogenada adenina (A) pela timina (T), conforme a seqüência representada a seguir: |
||||||||||||||||||||||
|
||||||||||||||||||||||
As mutações que afetam o controle dos genes para as sínteses equilibradas entre as globinas alfa e não-alfa são muito diversificadas. Em geral, essas mutações provocam diminuições no volume de síntese de uma das globinas com intensidades variáveis, ou até mesmo ausência de síntese. Nesses casos ocorrem desequilíbrios do conteúdo entre a globina produzida normalmente e a alterada. Quando a mutação bloqueadora de síntese de globina atinge, por exemplo, o gene alfa (talassemia alfa), há diminuição do conteúdo de globina alfa dentro das células eritróides, enquanto a síntese de globina beta se faz normalmente. Essa diferença de síntese entre globinas alfa e beta promove o desequilíbrio entre elas, e as conseqüências fisiopatológicas são proporcionais ao tamanho do desbalanceamento ocorrido (figura 5.1). |
||||||||||||||||||||||
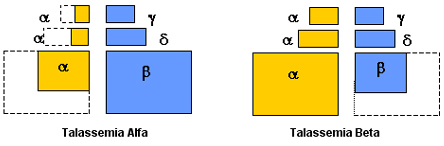 Figura 5.1 – Esquematização do equilíbrio entre globinas na formação de hemoglobinas normais, e dos desequilíbrios na talassemia alfa (diminuição da síntese de globina a) e na talassemia beta (diminuição da síntese de globina b). |
||||||||||||||||||||||
| Dessa forma, conceitualmente se denominam hemoglobinas variantes aquelas que apresentam estrutura química diferente à da sua hemoglobina normal correspondente (A, A2 ou Fetal), motivada pela mutação de uma ou mais bases nitrogenadas que resultam na substituição de um ou mais aminoácidos nas globinas alfa, beta, delta ou gama. As hemoglobinas anormais são aquelas consideradas variantes, bem como as hemoglobinas normais com alterações quantitativas, por exemplo: Hb A2 elevada, Hb Fetal elevada, Hb A2 diminuída. As talassemias consistem um conjunto de síndromes motivadas principalmente por alterações de sínteses quantitativas de globinas alfa e beta, causando desequilíbrio entre elas e variáveis graus de anemias hemolíticas, além de várias outras conseqüências patológicas. As hemoglobinopatias são designações destinadas às hemoglobinas variantes que causam anemia hemolítica, policitemia, cianose ou falcização, bem como as talassemias que serão apresentadas no item 7. Finalmente, há as hemoglobinas anormais e hemoglobinopatias não-hereditárias, que representam um grupo restrito de alterações das hemoglobinas normais causadas por agentes indutores e são também conhecidas como formas adquiridas. Entre as hemoglobinas anormais não-hereditárias destacam-se a Hb A2 diminuída nas ferropenias, a Hb A2 aumentada na malária, no diabetes e na doença de Chagas; a Hb Fetal elevada em certas doenças mielóides, em transplantados renais, em portadores de HIV ou por uso de determinadas drogas, e a Hb H adquirida nas doenças linfo e mieloproliferativas. Por outro lado, as hemoglobinopatias não-hereditárias mais comuns são as metaemoglobinas elevadas por indução de drogas oxidantes (sulfa e derivados, nitritos, anilina etc.), gases e solventes oxidantes. O grupo heme é tão importante quanto a globina por duas razões: como o agente que coloca o oxigênio à disposição da célula, que a ativa e transfere elétrons, e pelo fato de ser um pigmento corado que possibilita o estudo da diferenciação e maturação dos precursores eritrocitários. Assim, alterações das enzimas que participam das transformações dos compostos químicos envolvidos na síntese do anel de porfirina dão origem às porfírias e protoporfírias eritropoiéticas (ver figura 3.12 do ítem 3). |
||||||||||||||||||||||
 |
||||||||||||||||||||||
|
||||||||||||||||||||||