Autor: Paulo Cesar Naoum
O processo fisiológico da molécula
da hemoglobina obedece a um dinamismo que inclui a transformação
do seu estado oxigenado para a forma oxidada e vice-versa. Esse
desempenho é constante quando, em resposta à queda
na pressão de oxigênio, a oxiemoglobina é convertida
à hemoglobina oxidada e o oxigênio é liberado
aos tecidos. Esse estado de transferência é natural
e faz com que a hemoglobina seja continuamente oxidada in vivo
da forma ferrosa para a férrica, porém uma série
de eficientes mecanismos de proteção a reconvertem
de volta à oxiemoglobina (Fig. 12.1). Diariamente cerca de
1 a 3% de hemoglobina total circulante (ou oxiemoglobina) convertem-se
espontaneamente em metaemoglobina. Em pessoas normais, os níveis
quantitativos de metaemoglobina estão presentes no sangue
em valores variáveis de 0,3 a 4% do total de hemoglobina.
Assim, a metaemoglobinemia é a situação clínica
em que mais de 4% de hemoglobina foram oxidados para a forma férrica. |
||||||||||||||||||||||||||||||||||||||||||
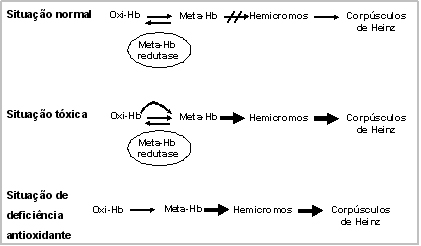
Figura 12.1 – Três situações específicas de geração de metaemoglobina. Na situação normal há equilíbrio entre oxi-Hb e meta-Hb, sem o desencadeamento da degradação em hemicromos e corpúsculos de Heinz. Nas outras duas situações o desequilíbrio entre oxi-Hb e meta-Hb, quer seja motivado por indução tóxica ou deficiência enzimática, promove a degradação da metaemoglobina em hemicromos e corpúsculos de Heinz. |
||||||||||||||||||||||||||||||||||||||||||
Causas da Metaemoglobinemia Metaemoglobinemia tóxica Muitas drogas são capazes de provocar metaemoglobinemia
tóxica como fenacetina, sulfas e derivados, bem como certos
compostos químicos: produtos de limpeza doméstica,
nitritos, nitratos e derivados do benzeno (nitrobenzeno e anilina).
As fontes de compostos químicos metaemoglobinizantes encontrados
na natureza são simples. Nitritos e nitratos podem estar
presentes em excesso, especialmente no espinafre e também
na água. No ar pode-se detectar óxido de nitrogênio
e de enxofre expelidos por indústrias de fertilizantes, de
refinamento de petróleo, entre outras. Todos esses produtos
causam oxidação da oxiemoglobina, transformando-a
em metaemoglobina. Explica-se esse fenômeno da seguinte forma:
a taxa de metaemoglobina se mantém normal quando a velocidade
de sua formação se iguala à de sua redução,
mediante o sistema de NADH-diaforase, ou por mecanismos enzimáticos
auxiliares. Na exposição de compostos tóxicos
ocorre um desequilíbrio nesse sistema, com aumento da velocidade
de formação de metaemoglobina e conseqüente elevação
da sua concentração (figura 12.1). Metaemoglobinemia por deficiência hereditária enzimática Esse estado pode suceder como resultado da deficiência
hereditária das enzimas de reconversão – as
meta-Hb redutases – caso em que é descrito
como “metaemoglobinemia congênita por deficiência
de NADH-diaforase”. Esse distúrbio caracteriza-se por
concentrações altas de metaemoglobina no sangue, na
ausência de administração de medicamentos ou
por exposição a compostos tóxicos oxidantes,
e sem ocorrência de qualquer anormalidade na parte globínica
da hemoglobina. Metaemoglobinemia por anormalidade molecular – Hemoglobinas M A cianose causada pelos dois grupos de metaemoglobinemias
abordados anteriormente pode ser tratada com agentes redutores,
tal como o ácido ascórbico, por via digestiva, ou
por injeção de azul de metileno. |
||||||||||||||||||||||||||||||||||||||||||
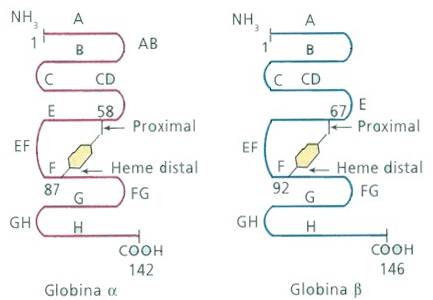
Figura 12.2 – Representação esquemática das globinas a e b e com as respectivas inserções do grupo heme. |
||||||||||||||||||||||||||||||||||||||||||
A hemoglobina M (Hb M) tem sido observada em todos os continentes. Frequentemente, os pais de pacientes portadores de Hb M são pessoas normais, Há, entretanto, relatos de Hb M em vários membros de uma mesma família. Na situação em que os pais não são portadores de Hb M, é bem possível que se trate de mutação recente (fresh mutation) do portador desta hemoglobina variante. Para que se possa entender a química e a fisiologia dos diferentes tipos de Hb M é importante recordar que o grupo heme está suspenso, no interior da subunidade molecular de globina, entre dois resíduos histidil. O ferro do grupo heme está ligado diretamente a uma histidina (proximal), enquanto o outro resíduo de histidina (distal) não está ligado quimicamente ao ferro, deixando, dessa forma, um espaço para o acesso do oxigênio durante a oxigenação do heme. Nesse contexto, cada molécula de hemoglobina abriga quatro grupos heme, sendo dois deles ligados às globinas a e outros dois às globinas b. Cinco hemoglobinas anormais, coletivamente denominadas hemoglobinas M, ocorrem por estar continuamente na forma de metaemoglobina e têm a substituição dos resíduos de histidinas distal ou proximal, das globinas a, b ou g (Tabela 12.1), e em razão disso, não se ligam ao oxigênio. |
||||||||||||||||||||||||||||||||||||||||||
Tabela 12.1 – Principais características laboratoriais das hemoglobinas M.
|
||||||||||||||||||||||||||||||||||||||||||
Das cinco hemoglobinas M, descritas até o presente, a Hb M Boston é a mais freqüente, sendo descrita em populações de vários países, inclusive o Brasil. Sob o ponto de vista clínico, há uma diferença entre a cianose causada pela Hb M nas crianças, o que permite prognosticar se a anormalidade da variante é de globina a ou b. Em recém-nascidos com Hb M, de anormalidade na cadeia a, a cianose é observada nos primeiros dias de vida. Esse fato ocorre em virtude da globina a se combinar com as globinas a, b e g, metaemoglobinizando as três hemoglobinas: A, A2 e Fetal, respectivamente. Entretanto, se a anormalidade é na globina b, a cianose não está visível até que boa parte de Hb Fetal tenha sido substituída pela Hb A, fenômeno que normalmente acontece a partir do segundo ou terceiro mês de vida. Nesses casos de Hb M com mutação de globina b, é comum ocorrer discreto grau de anemia hemolítica, associada a bilirrubinemia e icterícia. As hemoglobinas M, igualmente a outras variantes, são transmitidas como caráter co-dominante, com Hb A apresentando maior concentração. O estado de homozigose é incompatível com a vida. Metaemoglobinemia na doença falciforme A doença causada pelas células falciformes
se caracteriza por um conjunto de sinais e sintomas provocados pela
deformação de expressiva quantidade de eritrócitos
com hemoglobina S (Hb S) induzidos pela desoxigenação.
Esses eritrócitos com Hb S, quando oxigenados (oxi-Hb S),
se apresentam morfologicamente normais ou discóides no sangue;
às vezes, podem ser observados alguns eritrócitos
afoiçados ou falcizados. Entretanto, há situações
em que o processo de falcização é muito intenso
por causa da elevada concentração da Hb S desoxigenada
(desoxi-Hb S). A doença falciforme é um termo genérico
usado para determinar um grupo de alterações genéticas
caracterizadas pelo predomínio da Hb S. A introdução
da eletroforese de hemoglobinas como principal método de
investigação laboratorial contribui para entendimentos
clínico, genético e hematológico dos doentes
falcêmicos, pois sua aplicação permitiu explicar
que as diversidades clínica e hematológica estavam
relacionadas com a concentração da Hb S, bem como
sua associação com outras hemoglobinas. Assim, foram
caracterizados os principais genótipos que compõem
o grupo de doença falciforme e identificados por SS, SF (S/talassemia
b 0 e S/PHHF), SFA (S/talassemia
b +), SD, SC, SH (S/talassemia
a), entre outros. É evidente que
para o estabelecimento criterioso do genótipo da doença
falciforme é necessário que se façam análises
laboratoriais em familiares, especialmente nos pais, incluindo a
qualificação eletroforética em meios alcalino
e ácido, que comprovem a Hb S e suas associações
(SS, SD, SF, etc.), a quantificação densitométrica
da Hb S fracionada eletroforeticamente, avaliações
hematimétricas e morfológicas dos eritrócitos,
e a dosagem de Hb Fetal por metodologias bioquímica e imunológica. |
||||||||||||||||||||||||||||||||||||||||||
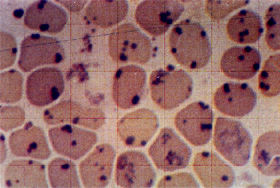
Figura 12.3 – Eritrócitos com precipitados de corpúsculos de Heinz após, após coloração específica com azul de cresil brilhante a 1%. |
||||||||||||||||||||||||||||||||||||||||||
Avaliação laboratorial das metaemoglobinemias As metaemoglobinemias são geralmente diagnosticadas
pelo médico, pois a própria história clínica
do paciente fornece indicativos da causa da cianose, por exemplo,
ingestão de sulfa, exposição a produtos químicos
oxidantes, entre outros. É comum, porém, o laboratorista
receber o material para quantificar e, até mesmo, qualificar
a metaemoglobina. A dosagem fornece o valor total de metaemoglobina
no sangue. A curva espectrofotométrica é importante
no fornecimento de subsídios qualitativos para se obter a
causa da metaemoglobina. Nos portadores de metaemoglobinemia tóxica
a curva é similar a da metaemoglobina A; na deficiência
enzimática é comum a observação de um
platô entre os comprimentos de onda 600 a 630, e naqueles
com metaemoglobinas M (Hb M), a absorção anormal de
alguns comprimentos de onda específicos indica o tipo de
Hb M (tabela 12.1). |
||||||||||||||||||||||||||||||||||||||||||
