Autores: ssssPaulo Cesar Naoum |
|||||||||||||||||||||||||||||||||||||||||||||||||||
As talassemias são um grupo heterogêneo
de doenças genéticas causadas pela redução
da síntese de globinas alfa e não-alfa (b,
d ou g).
Na realidade as formas mais comuns de talassemias se devem à
redução de globina alfa ou de globina beta (ver
figura 7.0) situações que originam as talassemias
alfa ou beta, respectivamente. Situações mais raras
envolvem a redução de síntese conjunta de
globinas delta e beta (talassemia d b),
ou de delta, beta e gama (talassemia d b g).
Em alguns casos de talassemias há redução
total de síntese de globina alfa ou de beta, caracterizando
as talassemias a 0 ou b
0, respectivamente, por outro lado quando a redução
de síntese é parcial denomina-se por talassemias
a + ou b
+. Pelo fato da talassemia beta, bem como as hemoglobinas
S, C e E serem as mais prevalentes respectivamente nos continentes
europeu, africano e asiático, não é raro
a ocorrência de interações entre talassemias
e essas hemoglobinas variantes: Hb S/Tal. b
0; Hb S/Tal. b +,
Hb C/Tal.b, Hb S/Tal.a
e Hb E/Tal.a. As combinações
entre genes talassêmicos com Hb S, principalmente, produzem
grande diversidade clínica dessa doença genética,
com variações que causam desde a morte fetal intra-útero
até situações assintomáticas. |
|||||||||||||||||||||||||||||||||||||||||||||||||||
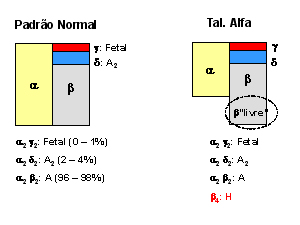
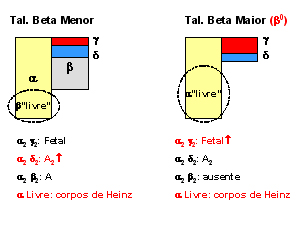
Figura 7.0 – Esquemas ilustrativos da síntese normal para hemoglobinas em comparação com talassemias alfa, beta menor e beta maior. |
|||||||||||||||||||||||||||||||||||||||||||||||||||
  |
|||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 7.1 - Classificação clínica das talassemias. |
|||||||||||||||||||||||||||||||||||||||||||||||||||
+: pouco; ++: moderado; +++: acentuado; ++++
muito acentuado; –: ausente. |
|||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||