Autor: Flávio Augusto Naoum |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Introdução A clínica da doença das células falciformes, notadamente da anemia falciforme, é muito variável pois depende de fatores genéticos, sociais, culturais e ambientais, principalmente.Nos últimos anos as expectativas com relação à morbidade e mortalidade da doença falciforme modificaram-se significativamente, em parte devido à maior precisão e precocidade no diagnóstico, e também pelo crescente volume de novos conhecimentos sobre a doença. A outra parte se deve à gradual sensibilidade dos órgãos de saúde pública em nosso país, motivado especialmente por movimentos sociais relacionados à população negra e às associações de portadores da falcemia. Apesar de todo o progresso que ocorreu nos últimos anos, o prognóstico do paciente com a doença falciforme permanece difícil de ser entendido devido à grande diversidade de manifestações clínicas, das variáveis que ocorrem entre diferentes faixas etárias, das condições sócio-econômicas, e do pronto atendimento. No Brasil, os pacientes com doença falciforme também padecem dessa expressiva heterogeneidade de patologias provenientes da falcização. Um dos primeiros trabalhos mais representativos sobre a história natural da anemia falciforme foi realizado em 1981 por Mara Hutz em pesquisa de prontuários de 409 pacientes com anemia falciforme cadastrados no Instituto de Hematologia Artur Siqueira Cavalcanti (atual HEMORIO) da cidade do Rio de Janeiro, de onde extraímos as principais manifestações clínicas expostas na tabela 6.8. É importante destacar que passados quase 30 anos muitas das informações relatadas, incluindo suas prevalências, mudaram. Essas mudanças ocorreram por duas razões principais: a primeira, pelo mérito de um trabalho de tal envergadura para aquela época, que mostrou o perfil do doente falcêmico e sensibilizou a comunidade científica para direcionar suas atenções a um problema de grande importância médica; a segunda, ocorreu pela significativa formação de profissionais das áreas médica e laboratorial que direcionaram seus estudos e estabeleceram critérios para o diagnóstico e conduta do paciente falcêmico. Esses fatos realmente causaram mudanças em todos os aspectos, e essas mudanças certamente foram notáveis. A seguir, apresentamos detalhadamente as manifestações clínicas mais comuns na doença falciforme e seus respectivos tratamentos. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.8 – Principais manifestações clínicas por faixa etária obtidas de 409 pacientes (200 homens e 209 mulheres) da região metropolitana do Rio de Janeiro (Profª. Mara Hutz, 1981). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(*) Para o cálculo da freqüência
foram utilizados, naturalmente, apenas os homens. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Grande parte da dor gerada nos episódios agudos é nociceptiva (transmitida por receptores da dor, os nociceptores) e resulta de estímulos somáticos ou viscerais. A dor somática é mais comum, geralmente intensa, localizada e lancinante, envolve inicialmente estruturas profundas como periósteo, medula óssea, articulações, músculos, tendões, ligamentos e artérias. Esta modalidade de dor, que pode ser focal ou referida, é transmitida por meio de fibras nervosas mielinizadas de condução rápida, principalmente fibras A d, as quais tem um limiar mais alto e por isso precisam de um estímulo forte, geralmente mecânico. A maioria destas fibras termina na lâmina I do corno dorsal da medula espinhal. A dor visceral está relacionada ao baço, fígado, pulmões, e outros órgãos; é geralmente vaga, mal localizada, difusa, continuada, e freqüentemente associada a náuseas, vômitos e sudorese. Esta modalidade é mediada por fibras não-mielinizadas de condução lenta, as fibras C, que podem transmitir estímulos mecânicos, térmicos e químicos, e são ativadas por inflamação, isquemia ou distensão. Estas fibras também entram na medula espinhal pelo corno dorsal, e terminam na lâmina II. A crise álgica é definida como grave quando da necessidade de assistência médica e analgesia parenteral com opióides durando pelo menos quatro horas. A ocorrência de três ou mais destas crises no doente ao longo de um ano denota doença falciforme de evolução grave. Platt e cols. realizaram estudo multicêntrico e prospectivo para determinar a apresentação das crises dolorosas em 3578 pacientes com doença falciforme. Os indivíduos com anemia falciforme (SS) e Sb 0 –talassemia foram mais freqüentemente acometidos, apresentando o dobro do número de crises que ocorreram na hemoglobinopatia SC e Sb + - talassemia. A variabilidade na incidência foi grande, e pôde ser apreciada na anemia falciforme, onde 40% dos pacientes não apresentaram dor ao passo que 5% evoluíram com 3 a 10 crises ao ano, e responderam por um terço do todas as crises observadas neste grupo. As crises dolorosas foram mais comuns na terceira e quarta décadas de vida, e a mortalidade foi maior nos adultos que pertenciam a esta faixa etária e evoluíram com 3 ou mais crises ao ano. Também foi observado que a frequência dos episódios variou diretamente com o valor hematócrito, provavelmente pela sua influência na viscosidade sangüínea, e inversamente com a concentração de Hb Fetal (Hb F). Haplótipos diferentes também têm sido implicados no quadro clínico do doente, por exemplo, pacientes com haplótipo do tipo Senegal parecem ter melhor evolução em relação àqueles com haplótipo do tipo Bantu. Além do episódio agudo, adultos com doença falciforme podem evoluir com dor crônica acompanhando quadro de úlceras de membros inferiores ou necrose avascular dos ossos (estes assuntos serão discutidos em outros tópicos). A investigação diagnóstica do doente com dor deve abranger história clínica com caracterização detalhada da dor (localização, intensidade, frequência, fatores predisponentes, sintomatologia associada, episódios e tratamentos prévios), exame físico, avaliação laboratorial e de imagem para definir a causa da dor e excluir outras possibilidades não relacionadas diretamente com a doença falciforme (DF). Radiografias dos locais envolvidos e avaliação mais específica com ressonância magnética podem auxiliar no diagnóstico de infarto medular e necrose avascular. O mecanismo pelo qual a dor se desenvolve e é percebida ainda não está completamente elucidado. Na DF a crise álgica é gerada pela oclusão microvascular e isquemia tecidual. Entre os fatores desencadeantes mais importantes estão a desidratação, hipóxia, infecção e acidose. Outras situações também podem precipitar o aparecimento das crises, como a mudança brusca de temperatura e o estresse emocional. Todavia, na maior parte dos casos, os episódios são imprevisíveis e não se consegue identificar nenhum fator predisponente. A oclusão microvascular acarreta lesão tecidual e esta, por sua vez, desencadeia uma resposta inflamatória com conseqüente liberação de citoquinas (interleucina-1) e outros mediadores inflamatórios. A interleucina-1 é um pirógeno endógeno com a capacidade da ativar o gene da ciclo-oxigenase para a produção de prostaglandina E2 e I2, as quais sintetizam terminações nervosas e facilitam a transmissão do estímulo doloroso ao córtex cerebral através da medula espinhal e do tálamo. Outros mediadores como bradicinina, histamina, K+ e H+ ativam as fibras nervosas aferentes nociceptivas provocando a resposta dolorosa. Além disso, os nociceptores ativados liberam substância P, que também facilita a propagação do estímulo doloroso e, juntamente com a bradicinina, causa vasodilatação e extravazamento de líquidos resultando em edema e dor local. Inibidores da dor como serotonina, encefalina, b-endorfina e dinorfina também interferem na percepção do estímulo doloroso. Deste modo, a intensidade da dor que o paciente sente é dependente da extensão da lesão e do equilíbrio entre seus ativadores e inibidores (figura 6.36). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
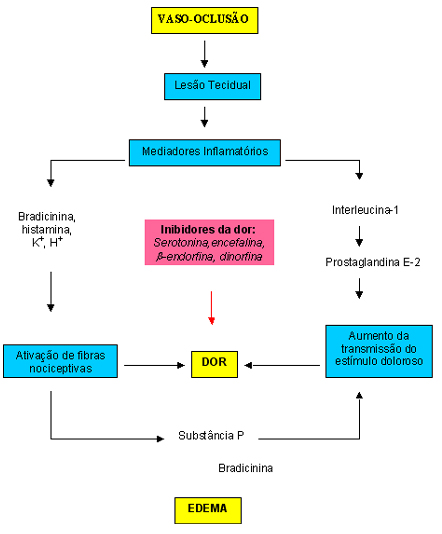
Figura 6.36 – Esquema caracterizando os principais fatores que causam as crises dolorosas. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Esta complexidade de fatores envolvendo o desenvolvimento e a sensação do estímulo doloroso é responsável pela grande variabilidade na apresentação das crises dolorosas observadas na doença falciforme. Além disso, a resposta inflamatória deflagrada pela lesão tecidual também leva ao aumento da atividade do sistema simpático com conseqüente aumento da isquemia tecidual, criando um ciclo vicioso da dor. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Via de regra, a conduta a ser
tomada frente a um paciente com crise dolorosa representa um problema
tanto do ponto de vista do médico quanto do paciente, e depende
da experiência prévia em relação ao uso
de analgésicos, enfoque psicológico, empatia e confiança
entre médico, paciente e familiares, estrutura e disponibilidade
dos serviços de atendimento, além de orientações
terapêuticas e preventivas levando em consideração
as condições econômicas, sociais e culturais
do paciente. Freqüentemente o portador de doença falciforme com dor é atendido nos serviços de emergência, onde a equipe médica e de enfermagem não estão familiarizados com o tratamento adequado da dor, e deixam de priorizar a queixa do doente, relevando a real dimensão do quadro doloroso. Este comportamento, por sua vez, aumenta a angústia do paciente – que conviveu por toda a vida com episódios dolorosos intensos e imprevisíveis acompanhados pelo medo da morte – , e reduz suas expectativas em relação ao tratamento, tornando o controle da dor cada vez mais difícil. A situação é ainda pior nos países pobres e em desenvolvimento, como no continente africano, onde a falta de recursos e medicações para o tratamento eficaz da dor, infra-estrutura deficiente para o atendimento do doente, e profissionais de saúde pouco qualificados, contribuem para maior morbidade e mortalidade decorrentes desta complicação. O tratamento analgésico freqüentemente envolve o uso de medicações diferentes, mas com ações sinérgicas. Assim é comum a associação de drogas não-opióides, como os anti-inflamatórios não esteroidais (AINE) que agem localmente bloqueando o processo inflamatório, opióides, os quais tem ação no sistema nervoso central, e ainda drogas adjuvantes, como anti-histamínicos e benzodiazepínicos (tabela 6.9). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 29 – Grupos de drogas utilizadas no tratamento da crise dolorosa. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Crises dolorosas de leve a moderada intensidade são geralmente tratadas ambulatorialmente com analgésicos não-opióides orais e, quando necessário, em associação com opióides fracos. Nos episódios graves os pacientes devem ser internados, recebendo preferencialmente uma abordagem multidisciplinar, e tratados de forma intensiva com opióides. A escolha do opióide e sua via de administração devem ser individualizadas, com especial atenção a esquemas bem sucedidos previamente usados pelo paciente. Entretanto o medo por parte do pronto-socorrista em relação aos efeitos colaterais como sedação e depressão respiratória, juntamente com a suspeita de dependência química pela droga, faz com que grande parte dos pacientes receba subdoses desta medicação em intervalos de tempo inadequados, prejudicando o alívio do quadro doloroso. Pacientes com doença falciforme são raramente dependentes químicos, todavia, o uso freqüente de opióides resulta em tolerância medicamentosa com a necessidade de doses cada vez maiores para se obter mesmo grau de analgesia. O problema da tolerância pode ser amenizado pela educação do paciente quanto à utilização domiciliar criteriosa do opióide, e usando-o em doses e intervalos adequados quando for indicado. Uma seqüência recomendável para o tratamento do paciente com dor pode ser apreciada na tabela 6.10. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.10 – Tratamento das crises dolorosas. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Preventivamente, bons resultados são obtidos com a utilização de hidroxiuréia, uma droga capaz de elevar a concentração de HbF no eritrócito, inibindo a polimerização e conseqüentemente a falcização eritrocitária. Este tratamento é indicado para os pacientes com anemia falciforme ou Sb-talassemia com três ou mais crises dolorosas graves ao ano, e reduz pela metade a incidência destas crises e de hospitalizações. O principal efeito colateral é mielotoxicidade, o que justifica cautela na dose prescrita e acompanhamento médico e laboratorial em intervalos de tempo menores. A eficácia desta droga em pacientes com hemoglobinopatia SC ainda não está totalmente estabelecida. O tratamento não farmacológico dos doentes inclui mudanças nos fatores ocupacionais, sociais e comportamentais que predispõem a ocorrência da crise álgica. Fisioterapia e terapia ocupacional são métodos capazes de melhorar a performance do doente e prevenir o desenvolvimento de complicações que causam dor crônica na doença falciforme. Muitos pacientes também se beneficiam de acompanhamento psicológico e discussão em grupo. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A síndrome torácica
aguda (STA) é uma complicação comum, de apresentação
variável entre os pacientes, e constitui atualmente a maior
causa de morte e a segunda maior causa de hospitalização
nos indivíduos com doença falciforme, nos países
desenvolvidos. É mais freqüente nas formas graves da
doença, como na anemia falciforme (ocorrendo em cerca de
um terço destes doentes) e na Sb
0 - talassemia. A STA é definida pela presença de febre, sintomas respiratórios (tosse, dor torácica e dispnéia) e infiltrados pulmonares na radiografia de tórax. Esta definição, apesar de abrangente, é útil para caracterizar uma síndrome grave muitas vezes negligenciada pelo socorrista, e que demanda cuidados específicos ao paciente com doença falciforme que se apresenta com sintomas respiratórios ou os desenvolve no curso de outra complicação, como crise dolorosa, e no pós-operatório. Os estudos de Vichinsky e cols. ofereceram contribuição valiosa para o entendimento da STA em relação à sua apresentação clinica e evolução, bem como na elucidação de suas causas. Nas crianças a incidência da síndrome é maior, principalmente naquelas entre 2 a 4 anos de idade. Os sintomas mais comuns nesta faixa etária são febre e tosse, raramente apresentam dor torácica, e os locais mais acometidos são os lobos superiores pulmonares. A STA na infância tem evolução mais branda, com menos necessidade transfusional, menor tempo de hospitalização e mortalidade, e está mais associada à infecção, com bacteremia acompanhando 14% dos episódios nas crianças com menos de 2 anos de idade, nas quais os germes mais freqüentemente identificados são Streptococcus pneumoniae e Haemofilus influenzae. Foi também observada variação sazonal na incidência desta complicação que foi maior nos meses de inverno, coincidindo com a época de maior ocorrência de infecções nas crianças, principalmente de vias aéreas superiores. No adulto, os sintomas mais freqüentes são dispnéia, calafrios e dor torácica importante, sendo que 18% desta população cursa com hipóxia. O acometimento pulmonar é geralmente multilobar e cerca de 50% dos episódios são precedidos ou acompanhados por crises dolorosas, o que reforça a hipótese de um maior componente vaso-oclusivo com embolia gordurosa no desenvolvimento da síndrome nestes casos. A frequência da STA no adulto é menor, no entanto, a evolução é mais grave e comprovada pela mortalidade que é quatro vezes maior em relação àquela observada na infância. Manifestações neurológicas ocorrem em 22% dos doentes e cerca de metade destes evoluem com insuficiência respiratória. Em 82% dos pacientes que têm STA o episódio é único, e um terço deles apresentam exame físico e ausculta pulmonar normal à apresentação. As alterações laboratoriais freqüentemente associadas ao quadro incluem queda do valor basal de hemoglobina e aumento do número de leucócitos. O tempo médio de internação dos doentes varia de 7 a 10 dias e cerca de 13% dos internados necessitam de ventilação mecânica (81% destes se recuperam). Alguns fatores têm sido implicados ao maior tempo de hospitalização, são eles: idade avançada, crise dolorosa nos membros à apresentação, febre, plaquetopenia, alteração radiológica extensa, terapia transfusional e insuficiência respiratória. O desenvolvimento desta complicação no pós-operatório está relacionado à diminuição da oxigenação e distúrbio de ventilação-perfusão resultantes do procedimento anestésico, com aumento do risco de infarto pulmonar ou infecção. As causas da STA, até então pouco esclarecidas, começaram a ser melhor estudadas nos últimos anos. Provavelmente a etiologia do evento é multifatorial, envolvendo infarto e infecção pulmonar, atelectasias secundárias a infarto de costelas e respiração superficial por dor, embolia pulmonar (tromboembolia ou embolia gordurosa) e trombose microvascular “in situ” devido à aderência dos eritrócitos ao endotélio (figura 6.37). O emprego do lavado bronco-alveolar para estudo etiológico é de grande valia na identificação de possíveis germes causadores das infecções que acompanham ou desencadeiam a STA e na confirmação de embolia gordurosa pelo achado de macrófagos carregados de gordura no material. Com este método, é possível especificar a causa em 40% dos casos (30% infecção e 10% embolia gordurosa). Além do pneumococo e H.Influenzae, outros agentes freqüentemente identificados são Clamídia pneumoniae e Micoplasma pneumoniae, seguidos por vírus sincicial respiratório e, em pequena fração dos pacientes, parvovírus B19. Recentemente, altas concentrações de fosfolipase A2 foram encontradas em indivíduos com STA; esta enzima degrada a gordura embolizada e libera ácidos graxos livres que causam lesões no parênquima pulmonar. Este fato sugere que a elevação da fosfolipase A2 está relacionada à ocorrência de STA decorrente de embolia gordurosa. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Figura 6.37 - Infartos ósseos em arcos costais. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Portanto, a abordagem diagnóstica inicial dos pacientes para STA deve ser realizada com radiografia de tórax, hemocultura e cultura de secreções respiratórias (preferencialmente lavado bronco-alveolar), gasometria, hemograma completo, cintilografia pulmonar de ventilação-perfusão (nos pacientes sintomáticos, mas com radiografia de tórax normal), e exclusão de trombose de membros inferiores. Com relação à fisiopatologia da STA, é preciso entender o comportamento do eritrócito falciforme no microambiente pulmonar bem como a influência de fatores agravantes decorrentes do comprometimento sistêmico pela doença falciforme. Os eritrócitos que chegam à circulação pulmonar estão desoxigenados e provavelmente contendo polímeros de HbS. Assim, qualquer doença pulmonar ou estado hipoventilatório que provoque hipóxia, deflagra a polimerização intracelular e a falcização eritrocitária, contribuindo para o fenômeno vaso-oclusivo. Em condições normais, a microvasculatura pulmonar tem a característica de reagir à situações de hipóxia com vasoconstrição para sustentar a relação ventilação-perfusão (V/Q) e manter a oxigenação no pulmão. Entretanto, na doença falciforme, tanto a hipóxia quanto a vasoconstrição favorecem não só a polimerização da HbS como também diminuem a velocidade de trânsito capilar dos eritrócitos e aumentam sua adesão ao endotélio. O aumento da adesão endotelial resulta do aumento na síntese da molécula de adesão VCAM-1 e da diminuição da produção do seu inibidor, o óxido nítrico (NOx). Participam do mecanismo de adesão os receptores eritrocitários (CD36 e a 4 b 1 integrina), os receptores endoteliais (CD36, a Vb 3 integrina, glicoproteina Ib-IX-V e VCAM-1) e ligantes presentes no plasma (trombospondina e fator de von Willebrandt). Por outro lado, na vigência de anemia, a vasoconstrição pulmonar secundária à hipóxia não ocorre adequadamente em virtude do acúmulo de óxido nítrico, apesar de sua baixa produção, resultante da sua diminuta captação pelos poucos eritrócitos circulantes. Esta condição promove o desequilíbrio da relação V/Q, o qual é agravado pelas atelectasias decorrentes de infartos costais e vertebrais que aumentam as áreas de “shunt”, ou seja, regiões perfundidas mas não ventiladas no pulmão. Com isso, há menos oxigenação, maior dessaturação da hemoglobina e hipóxia, formando o ciclo vicioso que envolve a STA (figura 6.38). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
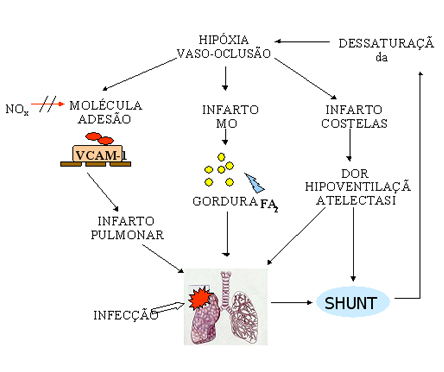 NOx-óxido nítrico; FA2-fosfolipase
A2, MO: medula óssea
NOx-óxido nítrico; FA2-fosfolipase
A2, MO: medula óssea Figura 6.38 – Fisiopatologia da Síndrome Torácica Aguda. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O tratamento da STA é
abrangente e enfoca os vários aspectos envolvidos na patogênese
da doença. Inicialmente, nos pacientes com dor torácica, recomenda-se analgesia adequada com emprego de opióides, o suficiente para evitar respiração superficial e conseqüentes atelectasias, porém com cautela pelo risco de depressão respiratória provocada por estas drogas. O emprego da espirometria, preventivamente, tem se mostrado muito eficaz na melhora do padrão respiratório dos pacientes, podendo até mesmo evitar o desenvolvimento da STA. Do mesmo modo, o tratamento intensivo com broncodilatadores traz bons resultados. A hidratação deve ser cuidadosa para evitar congestão e edema pulmonar, e não deve exceder o total de perdas e manutenção dos líquidos corporais. Oxigenoterapia é útil nos pacientes com hipóxia, e estes devem receber monitorização freqüente e assistência ventilatória adequada em unidade de terapia intensiva quando não houver resposta. Mesmo que infecção não seja documentada na maioria dos casos, a antibioticoterapia endovenosa é geralmente instituída, principalmente na vigência de febre, e o esquema proposto deve conter antibiótico da classe dos macrolídeos para cobertura das infecções causadas por Clamydia e Mycoplasma. A terapia transfusional tem por objetivo aumentar o valor da hemoglobina e reduzir a concentração da HbS. Tanto a transfusão simples como a ex-sangüíneo transfusão, para pacientes com hematócrito superior a 30%, estão indicadas quando há piora do padrão respiratório e hipóxia (PaO2<70mmHg em ar ambiente ou queda de 10% do valor basal em pacientes com hipoxemia crônica) e são úteis para evitar a progressão da STA e insuficiência respiratória, principalmente se administradas precocemente. Cuidados devem ser tomados em relação a situações que atrasam a indicação transfusional nesses doentes como confusão mental atribuída ao uso de narcóticos, edema pulmonar resultante de excesso de hidratação, mudanças freqüentes no antibiótico na falta de resposta clínica, quando na verdade todos estes sinais podem ser indícios da progressão da STA. Outras modalidades terapêuticas foram recentemente estudadas e podem beneficiar estes doentes. Dentre estas, o tratamento com hidroxiuréia mostrou eficácia em diminuir a frequência dos episódios de STA. O uso de dexametasona (0,3mg/Kg a cada 12h no total de 4 doses) reduziu o tempo de hospitalização, duração da febre, necessidade transfusional e dose dos opióides. Estudos experimentais utilizando a inalação de óxido nítrico no tratamento da STA evidenciaram redução na pressão da artéria pulmonar, melhora da relação ventilação-perfusão e da oxigenação pulmonar; no entanto, estudos controlados serão necessários para estabelecer o papel do óxido nítrico inalatório na STA. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O AVC é uma complicação
catastrófica da doença falciforme, e uma das maiores
causas de morte em crianças e adultos. Ohene-Frempong e cols.(1998)
realizaram um estudo prospectivo e multicêntrico com 4.082
pacientes e observaram que prevalência desta complicação
na doença falciforme foi de 3,7%, sendo o maior índice
encontrado nos pacientes com HbSS (4,0%), seguido pela Sb
0-talassemia (2,4%), Sb +-
talassemia (1,3%) e Hb SC (0,8%). Não houve diferença
entre os sexos masculino e feminino, quanto à prevalência.
A incidência documentada foi de 0,46 por 100 pacientes/ano
na doença falciforme. Nos pacientes com HbSS a incidência
situou-se em torno de 0,61 por 100 pacientes/ano, sendo mais freqüente
nos indivíduos com 1 a 9 anos de idade (0,84 por 100 pacientes/ano).
Mais da metade dos AVCs são isquêmicos (55%), seguido pelos hemorrágicos (cerca de 35%) e pelos acidentes isquêmicos transitórios (AIT – 10%). O AVC isquêmico é mais freqüente em pacientes com menos de 20 anos de idade, e o hemorrágico – responsável por alta taxa de mortalidade (24% nas 2 primeiras semanas após o evento) - tem sua maior incidência dos 20 aos 29 anos de idade. Os pacientes com doença falciforme que apresentam um episódio de AVC, têm um alto índice de recorrência, a qual pode ser diminuída, mas não abolida, com um programa de transfusão crônica. Cerca de dois terços das crianças com AVC isquêmico (AVCI), que não são transfundidas, podem apresentar um novo episódio dentro de três anos. Os fatores de risco relatados para AVCI são AIT prévio (principalmente), baixo valor de hemoglobina, episódio recente de STA, alta frequência de STA e pressão arterial sistólica elevada. O AVC hemorrágico (AVCH) está associado com baixo valor de hemoglobina e alto número de leucócitos. O efeito da concomitância da talassemia alfa na incidência do AVC na doença falciforme é controverso, mas é provável que haja proteção em relação à ocorrência do AVC, possivelmente devido à melhora do valor da hemoglobina. Esta complicação na doença falciforme acomete tanto vasos de pequeno quanto os de grande calibre. Infartos nas zonas marginais das artérias sugerem perda de vasos distais menores. Artérias maiores como as do polígono de Willis, carótidas internas e do sistema vértebro-basilar geralmente se apresentam com estenose e oclusão. Como conseqüência da oclusão dos vasos intracranianos maiores, há formação de uma massa composta de pequenos vasos sangüíneos friáveis, o chamado padrão “moya-moya”, freqüentemente observado nos pacientes com doença falciforme. Microscopicamente, a vasculopatia é caracterizada por hiperplasia da camada íntima com oclusão da luz do vaso, além de fragmentação e duplicação da camada elástica. Alguns vasos, principalmente no polígono de Willis, formam aneurismas, que podem se romper e provocar hemorragia. O AVC hemorrágico também pode ser o resultado da ruptura dos vasos “moya-moya”. A hemorragia, quando ocorre, é geralmente subaracnóide, mas hemorragia intra-ventricular e parenquimatosa também são observadas em alguns casos. A fisiopatologia do AVC na doença falciforme é pouco conhecida, e a sua etiologia é provavelmente multifatorial. Ao contrário de outros leitos vasculares, a oclusão com conseqüente infarto no AVC parece ocorrer mais freqüentemente nos vasos cerebrais maiores (principalmente artéria cerebral média e carótida interna) do que na microvasculatura. Em adição à falcização do eritrócito, outros fatores ou mecanismos patogênicos são provavelmente operantes no subgrupo de pacientes com doença falciforme que desenvolvem AVC. É possível a existência de um estado hipercoagulável nos doentes falciformes. A hiperhomocisteinemia está associada ao o risco de doença cerebrovascular e outros problemas vasculares na população geral. Houston e cols. estudaram 100 pacientes com doença falciforme, incluindo 16 com AVC, e observaram que os níveis de homocisteína estavam correlacionados com a ocorrência de AVC, onde os pacientes com níveis de homocisteína acima da média tiveram probabilidade 3,5 vezes maior de terem desenvolvido AVC. O mecanismo pelo qual a homocisteína aumenta o risco de doença vascular é desconhecido, mas têm-se demonstrado que ela inibe a expressão da trombomodulina, uma glicoproteína anticoagulante que age como cofator da trombina, ativando a proteína C. Esta redução na atividade da proteína C poderia predispor estes pacientes à trombose. Além disso, Khanduri e cols. identificaram baixos valores de proteína C na doença falciforme, e uma diminuição significante desses valores no grupo de doentes que tiveram AVC. Deste modo, pode-se sugerir que um screening para trombofilia à época do diagnóstico ajudaria a identificar crianças com risco de desenvolverem AVC e que estudos devem ser desenvolvidos no sentido de determinar a importância da terapia com anticoagulantes em baixa dose na doença falciforme com AVC. Além da adesão aumentada dos eritrócitos falcizados ao endotélio vascular, a regulação anormal do tônus vasomotor também contribui para a vaso-oclusão, incluindo AVC, nos pacientes com doença falciforme. O óxido nítrico (NOx) é um importante regulador do tônus vascular normal, adesão celular, e trombose. Deste modo, French II e cols. estudaram a adesão de eritrócitos-SS e do NOx na microvasculatura cerebral de ratos, observando que os eritrócitos-SS têm maior adesão à microvasculatura cerebral em relação ao grupo controle, e que a combinação da inibição da síntese de NOx e infusão de eritrócitos falciformes predispuseram à interrupção do fluxo sangüíneo cerebral e morte. Além disso, têm-se demonstrado que os níveis de NOx podem estar diminuídos localmente ou sistemicamente na doença falciforme, e recentemente, que leucócitos de pacientes com doença falciforme liberam grande quantidade de íon superóxido, um conhecido carreador do NOx. Estes estudos apontam um potencial terapêutico para o tratamento dos pacientes com doença falciforme com NOx. O quadro clínico inicial é semelhante ao da população geral e caracteriza-se por hemiparesia aguda, afasia ou convulsões nos AVCs isquêmicos, e cefaléia intensa nos episódios hemorrágicos. Hemiparesia residual crônica, retardo mental e episódios convulsivos de difícil controle constituem seqüelas habituais. O diagnóstico diferencial de AVC frente a um paciente com sintomas neurológicos inclui encefalopatia, hipertensão intra-craniana, trauma craniano, meningite, distúrbio metabólico, neuropatia periférica, entre outros. Há vários métodos de imagem disponíveis para o estudo do ACV na doença falciforme. Técnicas como angiografia e angiorressonância são utilizadas para detectar doença vascular, sendo este último um procedimento não invasivo com boa sensibilidade em relação à angiografia standard. Tomografia computadorizada e ressonância magnética são usadas para identificar infartos e lesões isquêmicas. O ultrassom Doppler transcraniano é um método que analisa a velocidade do fluxo sangüíneo cerebral e, recentemente, vem se mostrando de grande valia na identificação de pacientes com alto risco de desenvolver AVC possibilitando, portanto, que estes indivíduos recebam tratamento preventivo. Reserva-se o termo “infarto silencioso” aos casos onde se detecta infarto do tecido cerebral nos exames de imagem, mas sem evidência de sintomas neurológicos. Todavia, crianças classificadas como portadoras de “infarto silencioso” apresentaram resultados inferiores em estudos neuropsicométricos e déficits neurocognitivos quando comparadas a crianças com ressonância magnética cerebral normal. Além disso, esta condição está relacionada a maior risco de ocorrência de AVC e maior extensão do acometimento neurológico. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O paciente com doença
falciforme que evolui com AVC deve ser abordado em dois momentos
diferentes. Inicialmente trata-se o episódio agudo e, a seguir,
o doente é direcionado a um tratamento à longo prazo
com regime de transfusão crônica, que embora eficiente,
apresenta controvérsias quanto a sua duração. No episódio agudo, deve-se descartar fenômeno hemorrágico, estabilizar os sinais vitais do paciente, instituir hidratação com cautela e realizar transfusão sangüínea. O método transfusional mais aconselhável nesta fase é a ex-sangüíneo transfusão (manual ou automatizada), pois ela ao mesmo tempo eleva o valor da hemoglobina total melhorando a oxigenação e perfusão tecidual, evita hiperviscosidade e reduz a concentração de HbS. Dada a alta taxa de recorrência após o primeiro episódio de AVC, faz-se necessário a instituição de uma terapia à longo prazo com finalidade preventiva. A estratégia mais eficiente no AVC isquêmico em crianças é a terapia transfusional crônica. O objetivo deste tratamento é manter a HbS abaixo de 30% nos primeiros 3 anos após o episódio isquêmico. Para tanto, é necessário que o paciente receba transfusões sangüíneas regulares em média 1 vez por mês. Após 3 anos, tolera-se manter a concentração da HbS abaixo de 50% se o paciente apresenta-se estável do ponto de vista neurológico. Nas crianças, a terapia transfusional crônica reduz a taxa de recorrência do AVC para menos de 10%. Em adultos este tratamento ainda não foi aplicado de forma regular, e sua eficácia permanece incerta nos episódios hemorrágicos. A duração da terapia transfusional crônica não está estabelecida. Tentativas de descontinuar o tratamento apresentaram resultados conflitantes, com recorrências ocorrendo em pacientes que descontinuaram o tratamento após 10 anos de transfusões regulares, e outros doentes evoluindo sem novas complicações neurológicas com interrupção após 6 anos de tratamento transfusional (tabela 6.11). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.11 – Evolução de pacientes com doença falciforme e antecedente de AVC, após interrupção do tratamento transfusional. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
*Wang e cols; 1991; **Rana e cols; 1997. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
No entanto, a melhor opção continua sendo o regime transfusional crônico prolongado e por tempo indeterminado, devido à menor probabilidade de recorrência nesta terapia. Deve-se ressaltar que apesar das vantagens em relação ao comprometimento neurológico, este programa transfusional à longo prazo é acompanhado pelos riscos de aloimunização, infecções e sobrecarga de ferro (hemossiderose), que dependendo da intensidade com que ocorrem, podem inviabilizar o tratamento transfusional. A hidroxiuréia pode ser uma alternativa ao regime transfusional para prevenir a recorrência do AVC. Ware e cols.(1999) observaram recidiva em apenas 19% dos pacientes que interromperam a terapia transfusional e iniciaram tratamento com hidroxiuréia. Deste modo, a hidroxiuréia parece ser uma opção para os pacientes que apresentam sérias complicações oriundas do regime transfusional crônico com necessidade de interrompê-lo. Entretanto, faltam estudos maiores e controlados para afirmar o real benefício deste tratamento alternativo. O tratamento do AVC hemorrágico é variável. Pacientes com hemorragia cerebral por ruptura de aneurisma geralmente realizam craniotomia com clipagem do aneurisma. A terapia transfusional crônica vem sendo empregada para crianças que apresentam AVC hemorrágico, mas em adultos esta prática ainda não está estabelecida. Acidente vascular cerebral e doença cerebrovascular são indicações para realização de transplante de medula óssea alogênico, uma modalidade terapêutica cada vez mais estudada na doença falciforme. De fato, pacientes com AVC que foram transplantados com sucesso apresentam estabilização do quadro neurológico e a grande maioria evolui sem eventos cerebrovasculares subseqüentes. A conduta no infarto silencioso não está definida, mas várias opções podem ser consideradas, como transplante de medula óssea não-mieloablativo (mini-transplante) com objetivo de obter quimerismo misto e estabilização clínica, terapia transfusional crônica ou hidroxiuréia. No entanto, a escolha destas opções depende da realização de estudos controlados envolvendo estes pacientes. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A única estratégia
para prevenir a ocorrência de um primeiro AVC na doença
falciforme é baseada nos resultados de um estudo randomizado
realizado por Adams e cols.(1998), o estudo STOP (stroke prevention
trial in sickle cell anemia). O método empregado para identificar
as crianças com risco de desenvolver AVC foi o ultrassom
Doppler transcraniano, capaz de detectar aumento na velocidade do
fluxo sangüíneo cerebral que ocorre em associação
com estenose e lesões obstrutivas dos vasos e, consequentemente,
denotam alto risco de acidente vascular cerebral. Foram estudadas
130 crianças com anemia falciforme (sem história de
AVC) que apresentavam alterações no estudo de Doppler
transcraniano (velocidade > 200cm/seg), submetendo 63 delas a
regime transfusional para manter HbS<30%, e 67 à terapia
de suporte clínico. Foram observados dez eventos isquêmicos
e um hemorrágico no grupo de suporte clínico, e um
evento isquêmico no grupo submetido à terapia transfusional,
com uma diferença de 92% no risco de AVC. Estes dados mostram
que as transfusões diminuem em grande escala o risco de um
primeiro AVC nos pacientes com anemia falciforme que apresentam
alterações no estudo do Ultrassom-Doppler. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O baço na doença
falciforme tem uma variedade de apresentações dependendo
da idade do paciente e do seu grau de doença. O ambiente
de hipóxia dos cordões de Billroth promove desoxigenação
e, conseqüentemente, falcização dos eritrócitos
nos pacientes com anemia falciforme (HbSS). As células falciformes
são rígidas, e perdem a plasticidade para penetrar
nos sinusóides esplênicos. No início da infância, a esplenomegalia é proeminente devido à congestão dos cordões de Billroth pelos eritrócitos falcizados. Embora o baço esteja aumentado, sua função é geralmente prejudicada, levando ao aparecimento dos corpos de Howell-Jolly (inclusões de fragmentos nucleares) no sangue periférico. Infartos esplênicos dolorosos ocorrem devido a empactação das células falciformes na microvasculatura. Crianças na primeira infância podem estar sujeitas a crises de seqüestro em decorrência do armazenamento (“pooling”) maciço e súbito dos eritrócitos no baço, resultando em rápido aumento do órgão associado a citopenias e necessidade de esplenectomia. Crises aplásticas e hemolíticas, geralmente associadas à infecção, são outras complicações que podem ocorrer. Cerca de 6% das crianças com anemia falciforme desenvolvem um estado de hiperesplenismo crônico, onde metade destes são precedidos por um episódio de seqüestro esplênico agudo que inesperadamente não se resolve. O baço permanece aumentado (>4cm do rebordo costal esquerdo), com níveis de Hb<6,5g/dl e plaquetas<200.000/mm3. Esplenomegalia é rara em adultos com doença falciforme devido à progressiva atrofia causada por infartos repetidos no baço, levando à fibrose do órgão. Os nódulos fibróticos são cobertos por ferro a cálcio, formando os corpos de Gamna-Gandy. Nos estágios avançados, o baço tem seu tamanho reduzido e torna-se fibrótico, resultando em auto-esplenectomia funcional. Entretanto, em outras situações como na doença da HbSC e na associação da HbS com talassemia, a esplenomegalia pode persistir até a idade adulta. Na doença da HbSC, a dilatação moderada do baço está presente em cerca de 2/3 das crianças e freqüentemente persiste na vida adulta. No entanto, a perfusão do baço está intacta e, como resultado, pode ocorrer infarto esplênico sintomático e seqüestro esplênico agudo em adultos, assim como em crianças. Apesar da preservação da perfusão esplênica, a função do baço está comprometida, e esta ocorre de forma mais gradual e numa idade mais avançada do que na anemia falciforme. Crises de seqüestro esplênico também podem ocorrer nestes pacientes, onde geralmente são brandas, auto-limitadas a não requerem tratamento com transfusão sangüínea ou esplenectomia. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O baço é o primeiro
órgão a sofrer com os efeitos da anemia falciforme
na microvasculatura. Durante a infância, o baço encontra-se
aumentado de tamanho em 75% dos pacientes, no entanto, a partir
dos 6 meses de idade inicia-se o processo de involução
que culmina na perda total da função esplênica
(asplenia funcional) em mais de 90% dos doentes no final da infância,
além da progressiva auto-esplenectomia que ocorre com o desenvolvimento
de fibrose local. Nos pacientes com HbF aumentada, o processo de atrofia esplênica é mais lento e a função esplênica é preservada por mais tempo. Estes indivíduos são, portanto, mais susceptíveis a complicações decorrentes da esplenomegalia como crises de seqüestro, infarto esplênico, hemorragia intra-esplênica, ruptura e abscessos. O seqüestro esplênico é uma complicação resultante da estase aguda dos eritrócitos falciformes nos sinusóides do baço, que aumenta de volume. Conseqüentemente, ocorrem anemia, reticulocitose, plaquetopenia leve e hipovolemia. Este evento é definido como uma queda súbita de pelo menos 20% do hematócrito basal, associada ao aumento de 2 cm ou mais do baço à palpação. A maior incidência ocorre entre 5 meses e 2 anos de idade. Os episódios estão comumente associados à infecções das vias aéreas superiores, mas a fisiopatologia não é bem esclarecida. O quadro clínico é caracterizado por palidez mucocutânea de instalação súbita, acompanhada de distensão e dor abdominal pela esplenomegalia, podendo ocorrer polidipsia. A hepatomegalia, por vezes observada, não é tão marcante quanto a esplenomegalia. A perda de volume sangüíneo no baço logo leva ao choque, com taquidispnéia intensa, taquicardia e astenia importante. Os episódios variam de intensidade, com predomínio das apresentações brandas que geralmente resolvem-se espontaneamente. Entretanto, episódios graves ocorrem e podem ser fatais se não tratados rapidamente. Pacientes adultos raramente apresentam seqüestro esplênico, os indivíduos mais afetados são aqueles com HbSC e Hb SS/a - talassemia. Na associação com a talassemia alfa, o envolvimento do pacientes com idade mais avançada pode ser explicado pela melhor reologia eritrocitária que acompanha esta condição, com conseqüente preservação da função esplênica até a idade adulta. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Por se tratar de complicação
possivelmente fatal, a crise de seqüestro esplênico deve
receber tratamento em caráter de urgência. A maior
parte da mortalidade nos casos decorre do choque hipovolêmico
e não da anemia, portanto a conduta inicial consiste em reposição
da volemia com administração de expansores de volume.
As transfusões de concentrados de hemácias ajudam a restaurar o volemia e a pressão arterial, no entanto há que se fazê-la com cautela, pois à medida que o processo se reverte com o tratamento transfusional, o baço começa a reduzir de tamanho liberando o volume sangüíneo que estava em seu interior, de modo que o valor da hemoglobina pós-transfusional é freqüentemente maior do que o esperado pelo montante transfundido. Deste modo, as transfusões devem ser feitas em pequenas alíquotas, o suficiente para estabelecer o equilíbrio hemodinâmico e, ao mesmo tempo, evitar hipervolemia. Quando a hemoglobina encontra-se abaixo de 5g/dl em vigência de seqüestro esplênico, recomenda-se que seja inicialmente transfundido um volume em ml/Kg igual ao valor observado da Hb. Por exemplo, se uma criança apresenta hemoglobina de 3g/dl, o volume transfusional inicial deve ser de 3ml/Kg. As transfusões subseqüentes devem ser indicadas pelo quadro clínico, e a quantidade estimada com base no valor da hemoglobina após a primeira transfusão. Pacientes que apresentam hipervolemia podem ser beneficiados pelo uso de diuréticos. A conduta à longo prazo para os indivíduos que apresentam episódios recorrentes de seqüestro esplênico ou hiperesplenismo grave inclui orientação e treinamento dos pais quanto à palpação do baço da criança, terapia transfusional crônica ou esplenectomia. Em relação à indicação da esplenectomia, deve-se levar em consideração a facilidade de acesso ao atendimento médico especializado por parte do paciente e a preservação da função esplênica. Quando há dificuldade para encontrar assistência especializada em tempo hábil, deve-se recomendar a esplenectomia após o primeiro episódio grave desta complicação. Nos pacientes onde o baço é funcional e o acesso ao serviço médico não é problemático, pode-se postergar o procedimento com terapia transfusional até os 3 anos de idade, com o intuito de protegê-las das crises de sequestro repetidas e manter a função esplênica no período de alta vulnerabilidade à infecções graves. Esplenectomia parcial pode ser também uma alternativa nestes casos. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A crise aplástica é
uma complicação aguda que ocorre principalmente em
crianças com doença falciforme e, fora das zonas endêmicas
de malária, é a maior causa de anemia aguda nesta
faixa etária. Esta queda abrupta no valor da hemoglobina
é ocasionada por aplasia ou hipoplasia eritróide transitória
em medula outrora hiper-regenerativa que, superposta à destruição
aumentada e continuada dos eritrócitos na periferia, provoca
anemia grave em poucos dias. A aplasia transitória está associada a infecções por vírus e bactérias, onde o agente etiológico mais freqüentemente identificado é o parvovírus B19. O parvovírus B19 ataca preferencialmente os precursores eritróides da medula ligando-se ao antígeno P, o qual funciona como receptor deste vírus na superfície dos eritroblastos. No entanto, nem todos os pacientes infectados por este agente desenvolvem crise aplástica. Além disso, os indivíduos que já tiveram contato com o vírus provavelmente desenvolvem imunidade por toda a vida. Deste modo, é importante determinar o estado sorológico do paciente, pois aqueles sem evidência de infecção ou sem IgG elevada para o parvovírus B19, devem merecer maior atenção e melhor investigação etiológica frente aos episódios de reticulocitopenia. O parvovírus B19 pode induzir crise aplástica e seqüestro esplênico. A crise aplástica deve ser suspeitada à apresentação de uma criança febril com piora da anemia, palidez e fraqueza progressiva, sem aumento importante da icterícia, e valores baixos de hemoglobina e reticulócitos em relação ao estado basal. A instalação do quadro é geralmente insidiosa, de modo que a queda do valor da hemoglobina, embora significativa, é bem tolerada pelo paciente. Em geral, somente a eritropoese é afetada, porém leucopenia e plaquetopenia podem ocorrer em alguns casos. O curso desta complicação é geralmente transitório durando de 1 a 2 semanas. Outra causa de hipoplasia eritróide transitória é o tratamento com oxigênio inalatório, onde os doentes podem evoluir também com reticulocitopenia. O quadro se resolve após a interrupção de tratamento inalatório. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Inicialmente, é necessário
conhecer o perfil sorológico do paciente para o parvovírus
B19. Crianças em avaliação por doença
febril, sob risco de parvovirose ou cujo perfil sorológico
para o vírus seja desconhecido, devem ser isoladas de gestantes
pelo risco da infecção em relação ao
feto. Também deve-se instruir os pais de crianças
com doença falciforme a procurar assistência médica
frente a um episódio febril, o que aumenta as chances do
diagnóstico precoce desta complicação. Pacientes que apresentam queda do valor basal da hemoglobina igual ou superior a 25%, reticulócitos diminuídos e sintomas decorrentes da anemia, devem receber transfusões de concentrados de hemácias para encurtar o período de anemia e os riscos decorrentes desta ao longo da duração da crise aplástica. As transfusões são preferencialmente realizadas em alíquotas, assim como no seqüestro esplênico, para evitar hipervolemia e hiperviscosidade sangüínea mediante a recuperação medular associada à resolução do quadro. Geralmente, os pacientes com aplasia eritróide transitória começam a apresentar eritroblastos no sangue periférico e aumento de reticulócitos dentro de 1 semana. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Entre as manifestações
decorrentes da vaso-oclusão na doença falciforme,
encontra-se o priapismo, definido como uma ereção
dolorosa, involuntária e sustentada do pênis, durando
mais de 30 minutos, associada ou não ao estímulo sexual. Normalmente a ereção é iniciada em resposta a estímulos psicológicos, táteis ou nervosos, e resulta do aumento do fluxo sangüíneo para ambos os corpos cavernosos e corpo esponjoso do pênis, os quais não possuem comunicação vascular entre si. Este influxo de sangue, mediado em parte pela diminuição da atividade a -adrenérgica e conseqüente vasodilatação arterial local, provoca ingurgitamento dos sinusóides os quais, distendidos, comprimem as veias contra a fáscia que reveste o órgão e dificultam a drenagem venosa causando tumescência peniana. Este processo é revertido através do aumento gradual da resposta a -adrenérgica, com vasoconstrição arteriolar e restabelecimento do retorno venoso. Na doença falciforme o problema está no processo de reversão da ereção devido à congestão dos corpos penianos pelas células falcizadas, mas o mecanismo pelo qual isto ocorre não está totalmente esclarecido. Outro fator que pode contribuir para o desenvolvimento desta complicação é o aumento local da oferta de óxido nítrico ao endotélio pela HbS, resultando em vasodilatação. Freqüentemente os corpos cavernosos são acometidos no priapismo, entretanto, alguns pacientes têm envolvimento dos corpos cavernosos e corpo esponjoso ao mesmo tempo. Cerca de 40% dos pacientes com anemia falciforme apresentam pelo menos um episódio de priapismo. Deve-se ressaltar que esta alta incidência é observada somente quando se questiona o paciente diretamente sobre o assunto, pois muitos desconhecem a natureza desta complicação e sua relação com a anemia falciforme, ou simplesmente se acanham em discutir espontaneamente o problema durante as consultas de rotina. A maior incidência de priapismo ocorre por volta dos 20 anos de idade, sendo infreqüente na primeira década de vida. Portadores de S/b -talassemia apresentam baixa incidência de priapismo; os indivíduos com hemoglobinopatia SC também são raramente acometidos. Laboratorialmente, esta condição está associada a baixos valores de HbF. Basicamente, a apresentação do priapismo pode ocorrer de duas formas: o episódio agudo recorrente e o episódio agudo prolongado. Os episódios agudos recorrentes são mais freqüentes, auto-limitados, com duração inferior a 3 horas, geralmente noturnos, provavelmente influenciados pela desidratação fisiológica e acidose metabólica (resultante da hipoventilação) que ocorrem durante o sono. Alguns fatores como relação sexual e ingestão de bebida alcoólica podem desencadear os episódios, no entanto, a ereção noturna espontânea parece ser o fator precipitante mais comum. A recorrência é altamente variável e pode suceder ao longo de anos. Caracteristicamente, a função sexual geralmente é mantida entre os episódios. O priapismo prolongado geralmente dura mais de 24 horas; trata-se de complicação grave com necessidade de internação, e freqüentemente é precedido por episódios agudos recorrentes, mas em alguns casos podem representar o único episódio de priapismo do paciente. Os episódios prolongados são acompanhados de dor intensa e raramente recorrem. A impotência sexual é a principal seqüela do priapismo e acontece em 1/4 dos casos, a grande maioria deles ocorrendo após episódios prolongados. Pacientes com recorrência freqüente de episódios agudos podem apresentar perda parcial da função sexual. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Devido à escassez de
estudos acerca do tratamento do priapismo na anemia falciforme,
ainda não há consenso sobre a melhor conduta ou seqüência
de medidas a serem tomadas nesta complicação. Os episódios auto-limitados geralmente são resolvidos com medidas simples como exercícios, banho frio, masturbação, analgesia ou hidratação. Na maioria das vezes não há necessidade de tratamento específico e os pacientes não procuram atendimento médico. Nas situações mais graves e prolongadas, com necessidade de internação, surgem as seguintes questões: até que ponto deve-se insistir no tratamento clínico? Qual o melhor momento para indicar uma conduta cirúrgica? Na verdade não existe uma resposta ou proposta objetiva para estas questões. Mas baseado na experiência de diversos centros e nos dados disponíveis na literatura, pode-se fazer algumas observações. Inicialmente, recomenda-se uma abordagem conservadora com analgesia adequada, hidratação e transfusões sanguíneas. Transfusões simples de concentrados de hemácias e eritrocitaférese para reduzir a concentração da HbS sempre fizeram parte do tratamento no priapismo, porém com eficácia variável. Além disso, a eritrocitaférese realizada para priapismo agudo vem sendo recentemente associada a manifestações neurológicas (síndrome ASPEN), o que tem limitado o seu uso. Quando não há resposta com o tratamento conservador, condutas invasivas devem ser adotadas. Geralmente esta decisão deve ser tomada até 12 horas da internação, evitando-se postergá-la por 24 a 48 horas porque isto prejudicaria os resultados do tratamento cirúrgico. Esta modalidade de tratamento deve ser precocemente indicada nos pacientes com antecedentes de outros episódios de priapismo, em vigência de um novo episódio mais grave e prolongado que os anteriores. O objetivo da conduta cirúrgica é retirar o sangue estagnado nos corpos cavernosos e prevenir recorrência à curto prazo. Nestes casos preconiza-se aspiração do corpo cavernoso, juntamente com irrigação ou administração local de drogas a-adrenérgicas. O sangue retirado pela aspiração é espesso e escurecido, podendo conter coágulos. Se ainda não houver melhora do quadro, pode-se realizar cirurgia para criação de uma fístula caverno-esponjosa. Esta técnica viabiliza um “shunt” entre a glande e o corpo cavernoso, permitindo a drenagem sangüínea dos corpos cavernosos para o corpo esponjoso (que não está afetado pelo priapismo). Tal procedimento não é recomendado nos casos onde o corpo esponjoso também está afetado. As complicações decorrentes desta cirurgia são freqüentes, com alta probabilidade de falha terapêutica. A hidroxiuréia, com eficácia comprovada nas crises dolorosas e STA pelo aumento da concentração de Hb F, pode reduzir a frequência dos episódios de priapismo, no entanto faltam estudos randomizados para comprovar seu benefício para esta complicação. O dietil-estilbestrol é uma droga anti-androgênica que, apesar dos poucos relatos na literatura, vem se mostrando eficaz no tratamento do priapismo, reduzindo sua frequência e duração. Recentemente Cançado e cols. descreveram bons resultados em pacientes com ataques freqüentes e refratários à conduta clínica, que obtiveram melhora importante quando tratados com dietil-estilbestrol. Preconiza-se a administração de 5mg ao dia até a melhora do quadro agudo, seguidos de manutenção com 2,5 mg ao dia, três vezes por semana por 2 a 4 semanas. Resultados colaterais incluem ginecomastia e disfunção erétil. Os doentes com impotência decorrente do priapismo podem ser beneficiados com colocação de próteses penianas e aconselhamento psicológico. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Da mesma forma que a osteonecrose,
as úlceras de perna são complicações
comuns, freqüentemente crônicas e incapacitantes, com
alta repercussão na qualidade de vida dos pacientes com doença
falciforme. No entanto, pouco se conhece em relação
à sua fisiopatologia e faltam estudos controlados para se
estabelecer o tratamento mais adequado dentre as várias opções
existentes até o momento. As úlceras de perna ocorrem uni ou bilateralmente, principalmente nos maléolos laterais e mediais dos tornozelos, mas podem se desenvolver no dorso dos pés e nas pernas, e existem raros relatos desta complicação nas mãos. Como as extremidades inferiores do corpo são locais expostos, traumas locais e picadas de inseto podem precipitar a formação das úlceras (figura 6.39). O exame físico das extremidades é essencial para detectar alterações na pele do paciente e úlceras em fase inicial. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
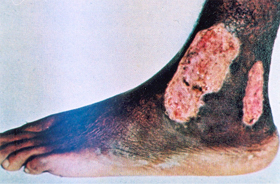
Figura 6.39 – Úlcera de perna. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Diversas são as opções
terapêuticas para as úlceras de perna (tabela 6.12). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.12 – Tratamento das úlceras de perna na doença falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Repouso e boa limpeza local para prevenir processos inflamatórios e infecciosos são as recomendações mais importantes para o sucesso do tratamento desta complicação, pois possibilitam a formação de tecido de granulação na superfície da úlcera favorecendo a sua reepitelização. Este tratamento local, principalmente a aplicação de compressas várias vezes ao dia, geralmente é suficiente para promover a cicatrização das úlceras menores (diâmetro inferior a 4 cm) em alguns meses. Ocasionalmente, há necessidade de realizar debridamento cirúrgico para limpar a base da úlcera, e facilitar sua resolução com o tratamento tópico. Apesar das culturas do local serem freqüentemente positivas – germes aeróbios e anaeróbios são identificados em mais da metade dos casos – isto não demanda necessariamente tratamento com antibióticos tópicos ou sistêmicos. No entanto, o aspecto da úlcera é fundamental para o diagnóstico de infecção e para decisão em relação ao tratamento. Assim, na presença de bordas hiperemiadas e secreção purulenta no local, que retardam a cicatrização, recomenda-se antibióticos tópicos (geralmente contendo aminoglicosídeos) e, se necessário, antibioticoterapia sistêmica baseada nos germes identificados pela cultura. Nas úlceras maiores (diâmetro superior a 8cm), de longa duração e cicatrização difícil, a terapia sempre oferece desafios e formas mais agressivas de tratamento devem ser propostas, como transfusões freqüentes, câmara hiperbárica e enxerto de pele. Apesar do tratamento transfusional, com elevação do valor da hemoglobina basal e redução da concentração da HbS, realmente auxilia na resolução do quadro, entretanto o seu uso é limitado pelas complicações conhecidas decorrentes da sobrecarga transfusional. O repouso prolongado é útil nestes casos porém, na prática, é freqüentemente inviável por parte do paciente. A realização do enxerto de pele é uma opção para úlceras grandes e não cicatrizadas. As condições para execução desta cirurgia são úlcera com base limpa, bom tecido de granulação e transfusão pré-operatória. No entanto, metade dos pacientes que realizam o enxerto apresenta recorrência, o que pode ser explicado pelo maior diâmetro, cronicidade e tempo de duração da úlcera no momento em que a cirurgia é indicada (tabela 6.13). Faltam estudos para determinar se o emprego precoce de tratamentos mais agressivos como este favoreceria o resultado do tratamento desta complicação. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.12 – Tratamento das úlceras de perna na doença falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
O tratamento com hidroxiuréia, e conseqüente elevação da hemoglobina total e Hb F, pode ser útil na prevenção do desenvolvimento ou da recorrência das úlceras de perna. Entretanto, os dados na literatura são conflitantes e, novamente, há necessidade de estudos controlados para determinar a sua importância. O uso prolongado da hidroxiuréia parece induzir a formação de úlceras em pacientes com doença mieloproliferativa crônica, porém este efeito não foi observado na doença falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Nos tecidos onde a circulação
sangüínea é lenta, como na medula óssea
e no baço, a desoxigenação e polimerização
da HbS ocorrem com maior facilidade, resultando em falcização
e oclusão vascular. Deste modo, depois do baço, o
osso é o principal órgão afetado pela doença
falciforme. Alterações ósseas decorrentes da hiperplasia medular crônica, que por sua vez resulta da hipersolicitação medular pela hemólise, são comumente encontradas nos pacientes. Isto ocorre porque os espaços medulares expandidos são preenchidos pela medula vermelha, que se espalha pelos canais haversianos alargados, invadindo a cortical e podendo-se estender até o periósteo. O defeito resultante deste fenômeno é o adelgaçamento da região cortical do osso, alargamento dos espaços medulares e trabeculação dispersa e irregular. No crânio observa-se proeminência da díploe, achatamento das lâminas externa e interna, e aumento da espessura dos ossos frontal e parietal. Além disso, trabeculação vertical ou perpendicular confere um aspecto radiológico de “terminação em cabelo” (hair-on-end) na díploe expandida. A protusão do maxilar superior leva a dentição defeituosa, caracterizada pela separação e angulação dos dentes incisivos, além de problemas para a oclusão da boca (gnatopatia da anemia falciforme). Os corpos vertebrais geralmente sofrem desmineralização pela hiperplasia medular, o que provoca achatamento e biconcavidade (vértebra em “H”). Também são achados freqüentes a osteoporose e a osteoesclerose das falanges terminais, entre outros. As lesões relacionadas principalmente ao fenômeno vaso-oclusivo, como a osteonecrose asséptica e a síndrome mão-pé, serão detalhadas a seguir. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A osteonecrose (ou necrose
isquêmica) é uma complicação comum, dolorosa
e debilitante da doença falciforme. Geralmente é insidiosa
e progressiva, acometendo principalmente quadril (cabeça
de fêmur) e ombros (cabeça de úmero), mas pode
afetar qualquer osso do corpo. Três quartos dos pacientes
com osteonecrose do úmero também apresentam lesão
no quadril. Por volta dos 35 anos de idade, metade dos pacientes
já apresenta evidências desta complicação.
Na infância a doença falciforme é a principal
causa de osteonecrose e, antes dos 15 anos de idade, a prevalência
é de 3% e a incidência de 2 casos por 100 pacientes/ano. No ombro, os sintomas de limitação do movimento estão ausentes em 80% dos casos no momento do diagnóstico. Por outro lado o envolvimento do quadril, de apresentação comumente insidiosa, pode ser agudo e simular uma artrite séptica ou sinovite. Com frequência, a manifestação desta complicação é bilateral, embora possa ocorrer em tempos e intensidades diferentes nos dois lados. O grau de osteonecrose é determinado pela classificação de Ficat, baseada em achados radiológicos, onde as lesões nos estágios iniciais são detectadas somente por ressonância magnética (tabela 6.15). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.15 - Classificação de Ficat para osteonecrose da cabeça do fêmur. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A prevalência, incidência, fatores de risco e evolução da necrose asséptica da cabeça do fêmur na doença falciforme foram determinadas em estudo prospectivo realizado por Milner e cols. envolvendo 2.590 pacientes com seguimento médio de 5,6 anos. A prevalência desta complicação em todo o grupo foi de 10%, sendo maior nos indivíduos SS e Sb 0-talassemia, e menor no subgrupo SC e Sb +-talassemia. Entre os pacientes SS, a prevalência foi maior naqueles com talassemia alfa concomitante. A incidência foi de 9% no grupo, e maior no subgrupo SS/a-talassemia, principalmente nos homozigotos para talassemia alfa (a-/a-), que apresentaram incidência de 4,4 casos por 100 pacientes/ano. Metade dos pacientes com diagnóstico radiológico de osteonecrose estavam assintomáticos, isto é, sem dor ou limitação de movimento. Os principais fatores de risco para o desenvolvimento desta complicação foram hematócrito alto, crises vaso-oclusivas freqüentes, VCM baixo, AST baixa. A correlação observada entre hematócrito e a incidência da osteonecrose decorre provavelmente do fato dos pacientes com SS/a-talassemia apresentarem hematócrito mais altos. A fisiopatologia desta lesão é pouco conhecida. Presume-se que o evento inicial seja a obstrução dos sinusóides medulares pelas células falcizadas, com conseqüente necrose da medula óssea e das células que formam o tecido ósseo. Esta necrose provocaria um processo de reparação óssea que apesar de melhorar a lesão, principalmente em indivíduos jovens, produziria também um aumento da pressão intramedular, o que levaria à reabsorção óssea e colapso da estrutura da cabeça do fêmur. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A artroplastia com colocação
de prótese é o principal tratamento para necrose asséptica
da cabeça do fêmur em estágios avançados,
mas o prognóstico na doença falciforme não
é tão favorável quanto ao observado nas artroplastias
para artrite de quadril por outras causas. Em geral, a colocação
de prótese no ato cirúrgico é dificultada pela
presença de esclerose óssea intensa. Cerca de 80%
dos doentes que operam têm menos de 35 anos e a necessidade
de revisão e reoperação da prótese pode
ocorrer em 30% dos casos após 4 anos. Após a cirurgia,
2/3 dos pacientes continuam com dor e 3/4 apresentam limitação
do movimento. Outras complicações relacionadas à
cirurgia também devem ser destacadas, como infecção,
fratura e síndrome torácica aguda. Como a osteonecrose é complicação que ocorre precocemente na vida dos indivíduos com DF, freqüentemente posta-se um dilema terapêutico entre o tratamento conservador que pouco garante em relação à resolução da dor, mas preserva em boa parte a mobilidade do membro, e o tratamento cirúrgico, mais eficaz quanto ao quadro doloroso, porém sujeito a várias complicações e limitação de movimento do membro afetado. O tratamento clínico conservador consiste em repouso, redução da sobrecarga de peso nas articulações de sustentação do corpo, administração de analgésicos e anti-inflamatórios não-esteroidais, hidroterapia. O tratamento fisioterápico propicia o fortalecimento da musculatura do quadril e da coxa, reduz o espasmo muscular local e auxilia na correção da postura. Nos estágios iniciais, bons resultados foram observados nos pacientes que foram submetidos à descompressão da cabeça do femoral, um procedimento cirúrgico que remove uma parte interna do osso e reduz a pressão no local. Os resultados desta modalidade de tratamento envolvem melhora clínica, com redução da dor e maior movimentação da articulação afetada, além da melhora radiológica observada em alguns casos. Apesar dos resultados animadores, há necessidade de um estudo controlado para determinar a importância deste procedimento no tratamento da osteonecrose na doença falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Também conhecida como
síndrome mão-pé, a dactilite é uma complicação
vaso-oclusiva aguda caracterizada por dor e edema no dorso das mãos
ou dos pés (ou ambos simultaneamente), por vezes acompanhados
de calor e eritema local (figura 6.40). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
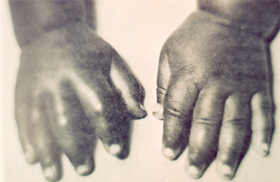
Figura 6.40 – Dactilite em criança com anemia falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As crianças são mais freqüentemente acometidas, principalmente entre 6 meses e 4 anos de idade. É comum a ocorrência da dactilite como a primeira manifestação da doença falciforme nas crianças. Em geral são episódios auto-limitados, durando de 1 a 2 semanas, podendo ser recorrentes, mas raramente deixam seqüelas articulares permanentes. Febre e leucocitose podem ser observadas na crise aguda e, nesses casos, deve-se fazer diagnóstico diferencial com osteomielite e artrite juvenil. Os achados radiológicos iniciais limitam-se à dilatação de partes moles, no entanto após 2 a 3 semanas podem surgir afinamento cortical, destruição dos metacarpos, metatarsos e falanges. Nos casos graves e repetitivos, a lesão acarreta crescimento desproporcional entre os dedos (figura 6.41). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
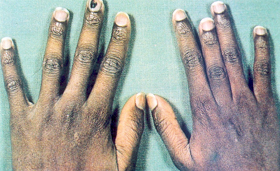
Figura 6.41 – Crescimento desproporcional entre os dedos da mão de paciente com anemia falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Uma vez que o quadro é auto-limitado na maioria das vezes, o tratamento se faz com cuidados locais e sintomáticos (analgésicos e anti-inflamatórios). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
A hiperosmolalidade, hipoxemia
e acidose local que caracterizam o micro-ambiente da medula renal
na doença falciforme, promovem a polimerização
da HbS e contribuem para a vaso-oclusão intra-renal, que
é o fenômeno responsável pelas complicações
deste órgão. Proteinúria ocorre em 26% dos pacientes e sua prevalência aumenta com a idade. É um sinal precoce no processo de lesão glomerular e sua fisiopatologia está associada à hipertrofia glomerular e esclerose glomerular focal. Outra manifestação que ocorre precocemente é a alteração do mecanismo renal para a concentração da urina. A lesão básica da hipostenúria é o fluxo sangüíneo prejudicado nos vasa recta, por estase ou obstrução permanente, que impede a manutenção do gradiente de salinidade normal na medula renal. A hematúria decorrente da doença falciforme resulta de necrose papilar (observada em 25% dos pacientes) ou da ruptura de vasos neo-formados dilatados. Geralmente é assintomática e tem evolução benigna, cessando espontaneamente. No entanto, cerca de 70% dos pacientes apresentam recorrência dos episódios de hematúria. A perda da função renal, em boa parte decorrente da hiperfiltração glomerular prolongada, aumenta consideravelmente a morbidade e a mortalidade dos pacientes, que sobrevivem em média apenas 4 anos após o diagnóstico da insuficiência renal. Comumente, uma queda nos valores basais de hemoglobina precede o diagnóstico desta complicação. Insuficiência renal aguda é pouco comum, na maioria dos casos está associada à hipovolemia (sem hipotensão ou sepse) e pode ser agravada pelo uso de anti-inflamatórios não esteroidais. Por outro lado, em estudo prospectivo conduzido por Powars e cols. a insuficiência renal crônica (IRC) foi observada em 4% dos pacientes com anemia falciforme e em 2% dos pacientes com HbSC. A incidência aumenta com a idade e acima dos 40 anos, 30% dos pacientes apresentam este diagnóstico. O haplótipo Bantu é o mais freqüente entre os indivíduos que desenvolvem insuficiência renal, talvez pela baixa concentração de HbF que apresentam. Já a associação com talassemia alfa previne ou retarda o desenvolvimento da glomerulopatia falciforme. A avaliação da creatinina deve ser cuidadosa e levar em consideração a influência da massa muscular diminuída e a alta taxa de filtração glomerular do paciente com doença falciforme, além da hiper-secreção tubular de creatinina que ocorre nesta doença. Portanto, valores de creatinina no limite superior da normalidade podem levar à suspeita de uma diminuição da função renal. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O uso de inibidores da enzima
de conversão da angiotensina-II (ECA) tem oferecido bons
resultados para a redução da proteinúria, além
do possível retardo na progressão da doença
renal nos pacientes. Cuidados como repouso e hidratação são suficientes para a resolução dos quadros de hematúria na maior parte dos casos. Em raras ocasiões, há necessidade transfusional pela perda acentuada de sangue. Desmopressina (DDAVP) e ácido epsilon aminocapróico também podem ser úteis no controle da hematúria. Com a IRC instalada, os pacientes entram em programa de hemodiálise crônica e tornam-se candidatos ao transplante renal, entretanto, o tratamento alternativo e precoce com hidroxiuréia e eritropoetina recombinante pode postergar a necessidade de tratamento dialítico e transfusões de sangue. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
As doenças do trato
biliar e do parênquima hepático constituem as principais
complicações do sistema digestório na doença
falciforme. Cerca de 70 a 80% dos pacientes com doença falciforme
apresentam hepatomegalia. Várias são as possíveis
causas de doença hepática nesta condição
(tabela 6.15). Além disso, é comum a presença
de mais de uma etiologia nas complicações hepáticas,
o que torna o difícil o diagnóstico diferencial entre
as causas. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.15 – Causas de doença hepática na doença falciforme. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Icterícia é freqüentemente observada nestes doentes e reflete a presença de hiperbilirrubinemia, principalmente às custas de bilirrubina indireta, decorrente da anemia hemolítica crônica. A intensidade da icterícia varia muito entre os doentes, e isto pode ser explicado por diferentes graus de comprometimento hepático, intensidades diferentes de hemólise e alterações genéticas do metabolismo da bilirrubina. De fato, a enzima UDP-glicuronil-transferase, responsável pela conjugação da bilirrubina, está aumentada na doença falciforme e impede que os pacientes evoluam com bilirrubinemia ainda mais elevada do que o usual. Assim, pacientes com doença falciforme que possuem alteração desta enzima, como os portadores da síndrome de Gilbert, terão um aumento isolado e expressivo da bilirrubina indireta, mas sem alteração da função hepática. Enzimas e testes de função hepática estão freqüentemente alterados, no entanto, estas alterações nem sempre traduzem existência de doença hepática. Deste modo, o aumento da fosfatase alcalina, principalmente em crianças e adolescentes, pode ocorrer devido ao crescimento ou lesões ósseas, e não por hepatopatia. Já a redução dos valores da proteína C e S (proteínas anti-coagulantes de produção hepática) comumente observada nestes pacientes é atribuída mais à disfunção hepática do que à coagulopatia de consumo. Algumas drogas têm seu metabolismo alterado pela disfunção hepática, como a morfina que passa a ter seu clearance aumentado. A biópsia hepática dos doentes revela achados histopatológicos compatíveis com a situação que levou à realização deste procedimento. Assim, nos indivíduos politransfundidos o principal achado é de hemossiderose, aqueles com hepatite viral apresentam sinais de hepatite crônica e cirrose, e nos que realizam biópsia por ocasião da colecistectomia observa-se dilatação sinusoidal e fibrose peri-sinusoidal. Síndrome do quadrante superior direito É uma síndrome caracterizada
por febre, icterícia e dor no hipocôndrio direito.
O diagnóstico diferencial deve ser feito entre crise dolorosa,
colecistite, seqüestro hepático agudo e crise hepática.
Dentre estes, o acometimento mais preocupante é a crise
hepática. A colelitíase é conseqüência
do metabolismo acelerado da bilirrubina decorrente da hemólise
crônica. Pode acometer crianças durante a primeira
década de vida e é observada em 75% dos adultos
com doença falciforme. A dor abdominal que acompanha o
quadro clínico de colecistite é difícil de
ser diferenciada de uma crise dolorosa e das várias outras
situações explanadas anteriormente que podem ocorrer
na doença falciforme. Dependendo da composição,
o cálculo biliar pode ser radiolucente ou radiopaco, assim
o método diagnóstico de escolha é a ultrassonografia.
A grande maioria dos pacientes com litíase biliar é
colecistectomizada de forma eletiva ou devido a um episódio
de colecistite aguda calculosa. A doença falciforme pode ocasionar lesões
oculares em diferentes graus de intensidade, geralmente resultantes
de fenômenos vaso-oclusivos, podendo por vezes passar desapercebida
ou cursar com complicações visuais, culminando com
cegueira. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 6.16 – Estágios da retinopatia falciforme proliferativa. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Inicialmente, ocorre a oclusão arteriolar na periferia da retina (Estágio I), com conseqüente dilatação destes vasos que estabelecem comunicações arterio-venosas entre a retina vascularizada e isquêmica (Estágio II). A seguir, formam-se neovasos a partir da superfície retiniana em direção ao vítreo e, por serem mal-formados, apresentam a forma de um leque, sendo chamados de “sea-fan” (molusco marinho com forma semelhante à dos vasos) configurando o Estágio III. Os vasos neo-formados são frágeis e permitem o extravazamento de sangue para o vítreo (Estágio IV), e a resolução e organização da hemorragia vítrea por meio da produção de tecido fibro-vascular promove tração na retina, provocando o seu descolamento (Estágio V). A retinopatia não proliferativa, ou de base, corresponde aos estágios I e II, enquanto que a retinopatia proliferativa corresponde aos estádios III, IV e V. A avaliação da retinopatia pode ser complicada pela possibilidade de regressão espontânea das lesões proliferativas, decorrentes de auto-infartos. Além da retina, alterações também podem ser observadas nos vasos da conjuntiva. Estas são mais comuns nos pacientes com HbSS e as modificações freqüentemente encontradas são aquelas que afetam a rede vascular superficial, e consistem na dilatação de segmentos dos capilares, que aparecem como vasos curtos, escuros, algumas vezes em forma de vírgula e geralmente sem conexões aparentes com o restante da rede vascular da conjuntiva. Estes achados são inversamente proporcionais ao valor de HbF e diretamente proporcionais ao número de células irreversivelmente falcizadas. Alguns pacientes podem apresentar dificuldade de adaptação visual no escuro devido a deficiência de zinco no organismo decorrente da alta excreção urinária deste elemento. A melhora clínica é obtida com a reposição de zinco. Tratamento O tratamento da retinopatia falciforme não
está definido. A infecção é a principal
causa de morbi-mortalidade durante os primeiros anos de vida na
doença falciforme, e o principal agente infeccioso nesta
condição é o Streptococos pneumomiae
(pneumococo). Antes da adoção de medidas preventivas
como triagem neonatal, vacinação contra pneumococo,
e instituição precoce de penicilina oral, as infecções
por S. pneumoniae constituíam a principal causa
de morte dos pacientes com menos de 20 anos de idade. Na África,
a situação é mais grave devido à malária,
que representa a causa mais freqüente de óbito nos
pacientes daquele continente. Além disso, os indivíduos
com doença falciforme apresentam perda da função
do baço precocemente. De fato, o baço tende a aumentar
seu volume até os 3 a 4 anos de idade quando passa a sofrer
regressão progressiva de tamanho. Entretanto, a função
do baço não se relaciona com o seu tamanho, assim,
mesmo que a criança tenha o baço aumentado, a função
deste já está comprometida (asplenia funcional). O alto risco de infecção e mortalidade
pelo S. pneumoniae em crianças com doença
falciforme levou à adoção de medidas extremamente
importantes. Tais medidas, como o screening neonatal,
tratamento profilático com penicilina oral e vacinação
contra pneumococo, quando instituídas precocemente, acarretaram
em redução significativa da frequência de
bacteremia. Assim como qualquer indivíduo com anemia
crônica, os pacientes com doença falciforme freqüentemente
apresentam sintomas cardiorespiratórios como palpitações,
dispnéia e fadiga, os quais exacerbam-se durante o esforço
físico. Além disso, a intensidade da anemia tem
relação com a capacidade de esforço físico
que encontra-se reduzida na metade dos adultos e em dois terços
das crianças. Ao exame físico, a ausculta cardíaca
raramente está normal, e os principais sinais encontrados
são sopro cardíaco, geralmente pansistólico
e mais audível em ápice, e cardiomegalia, detectada
ao exame clínico ou com auxílio de uma radiografia
de tórax e ecocardiograma. Sopros diastólicos são
incomuns e de significância clínica indeterminada,
já a presença de hipertensão pulmonar pode
gerar hiperfonese de 2ª bulha. Apesar da presença
de cardiomegalia e sopro cardíaco levantarem a suspeita
de insuficiência cardíaca congestiva, a contratilidade
cardíaca é freqüentemente preservada, o que
torna esta complicação incomum na doença
falciforme, principalmente entre as crianças. A insuficiência
cardíaca, quando presente, está geralmente relacionada
a eventos secundários como sobrecarga de volume e hipertensão,
que pode ser agravada pelo surgimento de lesão renal com
o avançar da idade, levando à hipertrofia ventricular
e insuficiência cardíaca. Hemossiderose é
a principal causa de insuficiência cardíaca na talassemia
maior, porém não é tão relevante na
anemia falciforme a ponto de causar cardiopatia, exceto em indivíduos
politransfundidos. Pacientes que desenvolvem insuficiência
cardíaca congestiva devem receber tratamento convencional.
Nos doentes gravemente afetados ou com angina pectoris,
é recomendável manter o valor da hemoglobina mais
alto que o habitual através de transfusões sanguíneas,
que devem ser realizadas cautelosamente para evitar sobrecarga
de volume e congestão pulmonar. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||