Autores:ssssssPaulo Cesar Naoum |
||||||||||||||||||||||||||||||||||||||
Introdução As técnicas utilizadas para avaliar amostras de sangue encaminhadas ao laboratório com suspeitas de doença falciforme incluem testes que podem ser subdivididos em quatro grupos: a) confirmatórios da presença de Hb S nos eritrócitos: teste de falcização ou teste de solubilidade; b) determinantes de genótipos (AS, SS, SC, etc.): eletroforeses de hemoglobinas em meios alcalino e ácido, isoeletrofocalização, dosagem de Hb Fetal, e cromatografia líquida de alta pressão (HPLC); c) determinantes de haplótipos por meio de técnicas de biologia molecular; d) monitoração: hemograma, contagem de reticulócitos, morfologia eritrocitária, dosagens de: ferritina, bilirrubina, ácido úrico, fosfatase alcalina, desidrogenase láctica (LDH) e metaemoglobina, e pesquisas intracelulares de corpos de Heinz, Hb H e Hb Fetal.Cada grupo de testes tem sua importância na informação de dados para fundamentar com segurança o diagnóstico completo da doença falciforme. Entretanto, para dimensionar sua importância no conjunto de informações, comentaremos cada um dos testes apresentados. Teste de falcização: é um teste de avaliação qualitativa que determina a presença ou ausência de Hb S nos eritrócitos. Seu princípio se baseia na indução da falcização por meio da desoxigenação da hemoglobina por drogas redutoras num microambiente formado no espaço entre lâmina e lamínula. Vários fatores interferem na sensibilidade e reprodutibilidade do teste, entre os quais se destacam a proporção entre os volumes de sangue e da droga redutora, falha na vedação do microambiente, e tempo de reação. O teste de falcização é o menos indicado devido ao baixo grau de resolução, pois quando positivo não diferencia o estado de associação genética (genótipo) da Hb S (Hb SS, Hb SF, Hb SC, Hb AS, Hb SD). Além disso, há muitos casos de "falso negativo", ou seja, eritrócitos com Hb S que submetidos ao teste de falcização não falcizam. Teste de solubilidade: esse teste é fundamentado no maior grau de insolubilidade da Hb S em comparação com a Hb A, em solução hipertônica contendo potente droga redutora (ditionito de sódio). Após misturar o sangue do paciente (suspeito de doença falciforme) com a solução hipertônica redutora, pode ocorrer ou não a turvação da mistura. A turvação indica insolubilidade da Hb S, enquanto que a transparência da mistura evidencia a ausência da Hb S. O teste tem baixa reprodutibilidade, podendo permanecer transparente mesmo na presença de Hb S. Também é um teste qualitativo, ou seja, a turvação ou insolubilidade da mistura não indica o tipo de associação da Hb S. Eletroforese alcalina de hemoglobina: a eletroforese alcalina é assim denominada devido ao fato do tampão utilizado para esse fim ter pH variável entre 8 e 9. Nessa faixa de pH a mutação que deu origem à Hb S (b 6 Glu ® Val) promoveu uma mudança de carga elétrica da molécula de Hb S, tornando-a menos negativa em relação à Hb A. Assim, quando amostras de sangue com diferentes genótipos são submetidas à eletroforese, a Hb S se move eletricamente de forma mais lenta que a Hb A (ver figura 5.5, capítulo 5 - Hemoglobinopatias). Há dois tipos de eletroforese alcalina de hemoglobina que são usados com maior frequência em laboratórios: o acetato de celulose e a agarose. Os dois testes apresentam excelentes padrões de qualidade técnica, permitindo a qualificação dos principais genótipos de doenças falciformes (figura 6.42), entretanto, como se pode ver pela figura 5.5 do capítulo 5 (Hemoglobinopatias) e da relação de hemoglobinas variantes que migram na mesma posição da Hb S (tabela 5.3, capítulo 5 de Hemoglobinopatias), é necessário um meio de comprovação de que a hemoglobina variante em questão seja realmente a Hb S. O melhor meio para comprovar é a eletroforese ácida de hemoglobinas. |
||||||||||||||||||||||||||||||||||||||
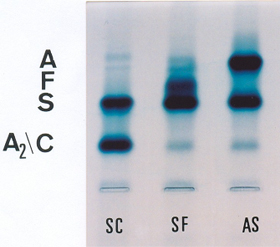
Figura 6.42 - Eletroforese alcalina de hemoglobinas em gel de agarose. Diferenciação da mobilidade eletroforética dos genótipos SC, SF e AS. Os traços de Hb A nos genótipos SC e SF se devem à sangue transfundido em ambos pacientes que cederam as amostras de sangue para análise. |
||||||||||||||||||||||||||||||||||||||
Eletroforese ácida de hemoglobina: essa eletroforese é sempre realizada em gel de agarose tamponada em solução de citrato ou fosfato com pH variável entre 5 e 6, por isso é também conhecida por agarose ácida. Embora o princípio básico seja a movimentação da molécula de hemoglobina induzida por sua carga elétrica (ou ponto isoelétrico), na realidade um outro fator importante interfere na mobilidade elétrica – a eletroendosmose. A eletroendosmose é um fenômeno físico-químico que se deve à interação elétrica entre as proteínas da agarose com as diferentes moléculas de hemoglobinas. É por essa razão que a Hb Fetal se move com maior rapidez que a Hb A na eletroforese ácida (figura 6.43), uma vez que na eletroforese alcalina a Hb Fetal é mais lenta que a Hb A. Mas o grande mérito da eletroforese ácida é diferenciar a Hb S das outras 22 principais hemoglobinas variantes que migram na mesma posição quando submetidas à eletroforese alcalina. A tabela 5.3 (capítulo 5 - Hemoglobinopatias) lista essas hemoglobinas variantes em relação à suas mobilidades na eletroforese em agarose ácida. Entre essas 22 hemoglobinas variantes que tem a mesma mobilidade da Hb S em eletroforese alcalina, a Hb D Los Angeles é a mais freqüente em nossa população. Assim, é comum o uso da expressão "diferenciar a Hb S da Hb D". Dessa forma, a aplicação conjunta das eletroforeses alcalina e ácida de hemoglobinas tem importante função no estabelecimento dos diferentes genótipos de hemoglobinas, conforme mostra a figura 5.5 do capítulo 5. |
||||||||||||||||||||||||||||||||||||||
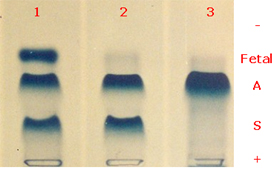
Figura 6.43 – Eletroforese ácida de hemoglobinas em gel de agarose. (1) Hb ASF em sangue de recém-nascido; (2) Hb AS; (3) Hb AA. Devido ao processo físico-químico de eletroendosmose a Hb Fetal migra com maior rapidez que a Hb A. |
||||||||||||||||||||||||||||||||||||||
Isoeletrofocalização: é também conhecida por eletroforese de focalização isoelétrica. Nesse tipo de eletroforese as proteínas são fracionadas de acordo com o seu pI ao longo de um gradiente de pH variável entre 5 e 14 que se forma durante a composição do gel. Este gradiente é estabelecido por um conjunto de anfólitos carregados isoeletricamente que permite identificar cerca de 80 tipos diferentes de hemoglobinas variantes. A IEF apresenta algumas vantagens sobre outros procedimentos eletroforéticos e cromatográficos. As vantagens em relação às eletroforeses se devem ao fato das excelentes resoluções obtidas para separar hemoglobinas variantes cujo pI difere entre 0,02 a 0,001. As vantagens sobre os procedimentos cromatográficos se revelam pela quantidade de amostras requeridas – 10 a 50m l na IEF, enquanto na cromatografia líquida de alta pressão (ou HPLC) são necessários 200 a 300m l, e principalmente por permitir a análise de várias amostras por vez (20 a 100 amostras). São essas razões que influenciaram o uso IEF no "screening" para Hb S em sangue de recém-nascidos. O fracionamento das hemoglobinas A, F, S, D e C por IEF tem alto nível de resolução, permitindo inclusive a diferenciação entre as Hb S e Hb D (figura 6.44). Devido ao alto custo operacional, seu uso tem sido restrito à pesquisa laboratorial de frações variantes, e em "screening" de hemoglobinas – onde o elevado número de análises por eletroforese oferece uma relação custo-benefício compensadora. |
||||||||||||||||||||||||||||||||||||||
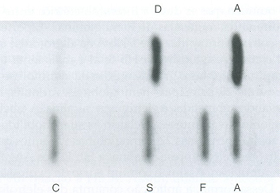
Figura 6.44 – Eletroforese de focalização isoelétrica (isoeletrofocalização) em gel de agarose. (1) "pool" de hemoglobinas A, F, S e C. (2) Hb AD. Por meio da isoeletrofocalização é possível obter fracionamentos de hemoglobinas com excelente nitidez, além de diferenciar as mobilidades das hemoglobinas S e D. |
||||||||||||||||||||||||||||||||||||||
| Dosagem de Hb Fetal: a determinação da concentração de Hb Fetal é fundamental em duas situações específicas: anemia falciforme e Hb S/talassemia beta, conforme mostra a tabela 6.1 (Introdução). Há uma relação direta entre concentração de Hb Fetal associada à Hb S, e o nível de hemoglobina total (g/dl) e o hematócrito. Da mesma forma, a elevação da concentração de Hb Fetal nos diferentes haplótipos de Hb S reduz a intensidade da gravidade clínica (tabela 6.3 da Introdução). A concentração de Hb Fetal é usualmente determinada por meio do teste bioquímico da resistência alcalina desta hemoglobina. Entretanto, há outras formas de avaliação quantitativa da Hb Fetal por metodologia imunológica e por densitometria do fracionamento obtido na eletroforese ácida de hemoglobina (figura 6.43). A metodologia imunológica tem boa sensibilidade e reprodutibilidade, porém o seu custo é alto. A densitometria da Hb Fetal dificilmente apresenta resultado coincidente com o obtido pela determinação bioquímica da resistência alcalina, isto se explica devido à densitometria ser um método essencialmente físico, enquanto a resistência alcalina é químico. A opção por um dos três métodos requer a padronização e obtenção dos valores de normalidade (média e desvio padrão). Cromatografia líquida de alta pressão (HPLC): é um processo de troca catiônica onde moléculas com cargas positivas são adsorvidas em uma fase estacionária da coluna cromatográfica, seguida por suas eluições induzidas pela passagem de um líquido (fase móvel) com altas concentrações de cátions. O eluato é detectado opticamente e quantificado computando a área do gráfico correspondente à fração eluída. A automação da HPLC promove esse processo com grande eficiência e precisão, permitindo a quantificação de hemoglobinas A, A2 e Fetal, bem como de hemoglobinas variantes. Assim, as hemoglobinas A, A2, Fetal, S, C, O Arábia, D Los Angeles, e G Filadélfia podem ser separadas uma das outras. Entretanto, seu uso deve ser sempre monitorado por eletroforeses alcalina e ácida, uma vez que há sobreposição de frações, como são os casos da Hb Lepore e Hb Korle-Bu que sobrepõe à Hb A2. Na HPLC a Hb A2 pode estar falsamente elevada na presença de Hb S. Biologia molecular: o agrupamento de genes da globina beta no cromossomo 11 humano é uma das regiões polimórficas mais bem estudada por técnicas de biologia molecular. A utilização de diferentes enzimas de restrição permite a ruptura de ligações entre determinadas seqüências de bases nitrogenadas, fato que determinou a qualificação dos cinco haplótipos (figura 6.9). Muitos estudos tem sido realizados com o objetivo de relacionar esses haplótipos com a diversidade clínica que se verifica entre pacientes com anemia falciforme (tabela 6.3). Por outro lado, a biologia molecular tem sido usada para a identificação pré-natal de anemia falciforme por meio da enzima Mst II que fragmenta o DNA da globina b S diferentemente da fragmentação do DNA da globina b A. Esses fragmentos submetidos à eletroforese de DNA em gel de agarose permite a separação do fragmento do DNA da globina b S (com 1,35 Kb) do DNA da globina b A (com 1,15 Kb). Uma outra aplicação da biologia molecular é usada na diferenciação da Hb S de outras hemoglobinas variantes que migram eletroforeticamente em posição similar à da Hb S, como são os casos da Hb D Los Angeles e Hb Korle-Bu. Nesses casos, o DNA extraído é amplificado e submetido a ação da digestão enzimática com diferentes enzimas de restrição. O material resultante da digestão é submetido à eletroforese em gel de agarose ou poliacrilamida que permite, após a revelação das bandas, a identificação da Hb variante. O hemograma: é uma análise que contempla diversas provas efetuadas, com a finalidade de avaliar quantitativa e qualitativamente os componentes celulares do sangue. Os itens avaliados incluem: contagem de eritrócitos, concentração da hemoglobina total, hematócrito, índices hematimétricos, leucócitos totais, contagem diferencial de leucócitos, plaquetas, e análises morfológicas de eritrócitos, leucócitos e plaquetas. A importância do hemograma nas doenças das células falciformes se destaca sob dois aspectos: no monitoramento hematológico do paciente e no auxílio do diagnóstico diferencial entre a anemia falciforme e a interação Hb S/talassemia. Assim, os índices VCM (volume corpuscular médio) e HCM (hemoglobina corpuscular média) tem grande importância na diferenciação entre Hb SS e Hb S/talassemia, conforme mostra a tabela 6.17. |
||||||||||||||||||||||||||||||||||||||
Tabela 6.17 – Diferenciação dos principais genótipos de Hb S relacionados aos valores de hemoglobina (Hb g/dl), volume corpuscular médio (VCM), reticulócitos e Hb Fetal. |
||||||||||||||||||||||||||||||||||||||
* Deleção de dois genes alfa ** Em eletroforese identificam-se as frações de Hb A, F e S; a Hb S tem concentração maior que a Hb A. |
||||||||||||||||||||||||||||||||||||||
Morfologia eritrocitária: é quase sempre muito diversificada pela presença de células falciformes, em alvo, dacriócitos, esquisócitos, eritroblastos, policromasia, corpos de Howell-Jolly, pontilhados basófilos, micrócitos e macrócitos, principalmente. Contagem de reticulócitos: a reticulocitose é comum nas doenças das células falciformes (tabela 6.17), e tem importância como marcador biológico do grau da eritropoiese. A pesquisa de corpos de Heinz na lâmina em que se avalia a contagem de reticulócito fornece subjetivamente o grau de oxidação da Hb S. Da mesma forma, utilizando a técnica da contagem de reticulócitos, é possível pesquisar a presença de precipitados intra-eritrocitários de Hb H para estabelecer a interação do genótipo de Hb S com talassemia alfa. Ferritina: é a mais importante proteína de reserva de ferro. É encontrada em todas as células, especialmente naquelas envolvidas na síntese de compostos férricos, no metabolismo e na reserva de ferro. A dosagem de ferritina é o mais fiel indicador da quantidade de ferro armazenada no organismo. Na carência de ferro, a ferritina diminui antes das alterações dos níveis de ferro sérico e das alterações da morfologia eritrocitária. Sua concentração está elevada em politransfundidos, nas anemias hemolíticas e megaloblásticas, e nas lesões hepáticas. Por fazer parte do grupo de proteínas da fase aguda, a ferritina se eleva em respostas às infecções, traumatismos e inflamações agudas. Muitos doentes falciformes são submetidos a constante reposição de concentrado de eritrócitos, fato que caracteriza o "regime" de politransfusão. Os eritrócitos transfundidos aumentam a sobrecarga de ferro e, assim, a dosagem de ferritina se apresenta como sensível marcador dessa situação. Bilirrubina: é o principal produto do metabolismo do grupo heme. Cerca de 70% da bilirrubina são provenientes da destruição de eritrócitos velhos, 15% provém de fontes hepáticas, e 15% se deve à destruição de eritrócitos defeituosos na medula óssea. Assim, no caso das doenças das células falciformes a excessiva quantidade de eritrócitos defeituosos induz sua destruição precoce e, consequentemente, eleva a concentração de bilirrubina indireta para valores acima de 5mg/dl. Ácido úrico: é o maior produto do catabolismo das purinas. Diversos fatores como dieta, predisposição genética, sexo, idade, peso, medicamentos, uso de álcool, e associação com outras patologias como diabetes mellitus e distúrbios lipídicos podem alterar os valores séricos e desencadear o desequilíbrio entre a absorção e a excreção de ácido úrico. A hiperuricemia ocorre quando a concentração de ácido úrico ultrapassa os valores de referência. Entre as várias causas que aumenta o ácido úrico no sangue destacam-se as anemias hemolíticas devido à reutilização de ácidos nucléicos provenientes da degradação da globina motivada pela precoce destruição dos eritrócitos, fato comum nas doenças das células falciformes. Portanto, nesses casos, a elevação do nível de ácido úrico é conseqüência da hemólise. Fosfatase alcalina: é uma enzima presente em praticamente todos os tecidos do organismo, e em especial nas membranas das células dos túbulos renais, ossos, placenta, trato intestinal e fígado. Assim, a fosfatase alcalina encontrada no soro é resultante da presença de diferentes isoenzimas provenientes de diferentes órgãos, porém com predomínio das frações ósseas e hepáticas. Na prática clínica, a grande utilidade está na investigação de doenças hepatobiliares e doenças ósseas que cursam com aumento da atividade osteoblástica. Na anemia falciforme é freqüente a ocorrência de cálculos biliares (colelitíase) devido à bilirrubinemia desencadeada pelas crises de hemólise, situações que elevam a concentração sérica de fosfatase alcalina. Desidrogenase láctica (LDH): é uma enzima intracelular que induz a oxidação reversa do lactato em piruvato. Está amplamente distribuída em todas as células do organismo, concentrando-se especialmente no miocárdio, rim, fígado, eritrócitos e músculos. A elevação da LDH está relacionada em todas as situações em que ocorre grande destruição celular, como são os casos das anemias hemolíticas e megaloblástica, insuficiência cardíaca congestiva, doenças musculares, lesões hepáticas, e neoplasias, entre outros. Nas doenças falciformes associadas à crises de hemólises, a LDH apresenta-se com concentrações elevadas, fato que a caracteriza como sensível marcador biológico da destruição de eritrócitos. Metaemoglobina: conforme foi apresentado no capítulo 5, a constante oxidação da hemoglobina eleva o nível de metaemoglobina. Quando a concentração da Hb S é maior que 50% a metaemoglobinemia desencadeia a degradação da Hb S com formação de corpos de Heinz. Todo esse processo provoca lesões na estrutura da membrana do eritrócito falciforme, induzindo sua destruição pelos macrófagos. Assim, a dosagem de metaemoglobina nas doenças das células falciformes se apresenta como importante indicador biológico relacionado à oxidação da Hb S. Pesquisas intracelulares de corpos de Heinz, Hb H e Hb Fetal: os corpos de Heinz são precipitados de globinas a ou b que se aderem à membrana do eritrócito sob a forma de estruturas circulares. Quando a proporção de eritrócitos com corpos de Heinz é maior que 1 para 1000 eritrócitos normais é indicativo de processos oxidativos da hemoglobina. Nas doenças das células falciformes é comum a presença elevada de corpos de Heinz em proporções variáveis entre 1:50 a 1:200. A pesquisa de Hb H intraeritrocitária é necessária quando se suspeita de talassemia alfa. A talassemia alfa de um gene afetado é comum na população brasileira, com prevalência entre 15 a 25%, e sua identificação se faz pela presença de traços de Hb H na eletroforese alcalina de hemoglobina em acetato de celulose e também por meio da precipitação intra-eritrocitária de Hb H com corante redox (azul de crezil brilhante ou violeta de metil). Por ser uma lesão em apenas um gene alfa, dos quatro existentes, é muito difícil estar associada com alterações hematológicas e clínicas. Por outro lado, quando a lesão envolve dois genes alfa é comum a anemia microcítica e hipocrômica de grau leve. A prevalência da alteração de dois genes alfa afetados na população brasileira é variável em 3 a 5%. Assim, a interação entre anemia falciforme (Hb SS) e talassemia alfa com um ou dois genes alfa afetados é comum em nossa população. As alterações hematológicas na interação de Hb SS/talassemia alfa são menores quando comparadas com a anemia falciforme "pura" (Hb SS) conforme mostra a tabela 6.17. Tecnicamente as pesquisas intracelulares de corpos de Heinz e de Hb H obedecem o mesmo procedimento de incubação de sangue total com azul de crezil brilhante, cujas diferenças morfológicas podem ser apreciadas nas figuras 6.45 e 6.46. A pesquisa intracelular de Hb Fetal se faz por meio da técnica de eluição ácida no esfregaço de sangue total, em que Hb A é retirada (ou eluída) dos eritrócitos, enquanto que a Hb Fetal permanece dentro da célula. Após contra-coloração com eritrosina, ou até mesmo com Giemsa, é possível determinar a forma de disposição dos eritrócitos contendo Hb Fetal. Quando não há Hb Fetal os eritrócitos apresentam-se desprovidos de hemoglobina (figura 6.47). Quando a distribuição é heterogênea, ou seja, presença concomitante de eritrócitos corados (indicativo de Hb Fetal) e vazios (indicativo de Hb A eluída), é sugestivo de talassemia menor (figura 6.48) ou maior (figura 6.49). Quando todos os eritrócitos apresentam-se corados homogeneamente se trata de persistência hereditária de Hb Fetal (PHHF). Há dois grupos específicos de genótipos de Hb S associados com Hb Fetal , quais sejam: Hb S / talassemia beta e Hb SS / PHHF. No primeiro grupo a associação é interativa entre o gene b S e o gene b Tal, com anemia microcítica e hipocrômica e anemia variável entre grave (Hb S/tal b 0) a moderada (Hb S/tal b +), conforme mostram a tabela 6.1 e tabela 6.17. No segundo grupo situa-se a associação entre a anemia falciforme com PHHF, onde a Hb Fetal elevada inibe o processo de polimerização da Hb S fato que favorece o desempenho fisiológico da oxigenação, diminuindo a intensidade da anemia, bem como das manifestações clínicas (ver tabela 6.1). |
||||||||||||||||||||||||||||||||||||||
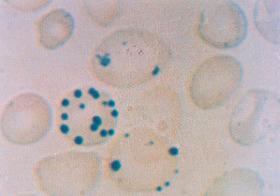
Figura 6.45 – Precipitados intra-eritrocitários de corpos de Heinz em pessoa com anemia falciforme. |
||||||||||||||||||||||||||||||||||||||
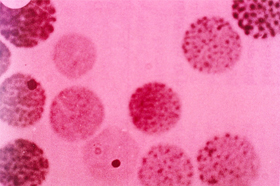
Figura 6.46 – Precipitados de Hb H em eritrócitos de pessoa com talassemia alfa. |
||||||||||||||||||||||||||||||||||||||
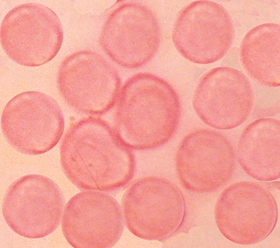
Figura 6.47 – Teste negativo para Hb Fetal intraeritrocitária. Os eritrócitos normais (Hb AA) tiveram suas moléculas de hemoglobinas eluídas após serem submetidos à técnica de eluição ácida. |
||||||||||||||||||||||||||||||||||||||
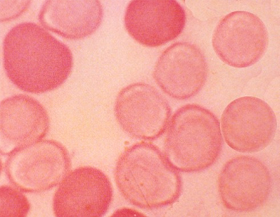
Figura 6.48 – Distribuição heterogênea de Hb Fetal intraeritrocitária na talassemia beta maior. Os poucos eritrócitos corados em escuro representam aqueles que não tiveram a Hb Fetal eluída após tratamento pela técnica de eluição ácida. Os eritrócitos desprovidos de coloração são eritrócitos com Hb A, cujas moléculas de hemoglobinas foram eluídas. Obs.: a dosagem bioquímica de Hb Fetal deste caso foi de 4,2%. |
||||||||||||||||||||||||||||||||||||||
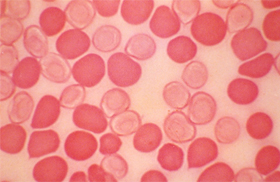
Figura 6.49 – Distribuição heterogênea de Hb Fetal intraeritrocitária na talassemia beta maior. 2/3 dos eritrócitos apresentam-se corados devido à presença de Hb Fetal que não foi eluída após tratamento com a técnica de eluição ácida. Obs.: a dosagem bioquímica de Hb Fetal deste caso foi de 68%. |
||||||||||||||||||||||||||||||||||||||
|
O traço falciforme caracteriza
o portador assintomático, laboratorialmente representado
pela associação das hemoglobinas A e S, ou Hb AS.
A concentração da Hb S no traço falciforme
é sempre menor que da Hb A, variando entre
30 e 40% (ver tabela 6.1 e figura 6.3 em Introdução).
Eventualmente, quando associado com anemia ferropriva ou talassemia
alfa, a concentração da Hb S pode se situar abaixo
de 30%. Pelo fato da menor concentração da Hb S em
relação à da Hb A, o portador heterozigoto
não padece da doença e não apresenta alterações
hematológicas. Os processos vaso-oclusivos sob condições fisiológicas normais inexistem, e, portanto, não há mortalidade e nem morbidades seletivas. Geralmente detecta-se o portador de Hb AS em estudos populacionais e pré-natal, ou em análises de familiares com membros portadores do gene da Hb S. No Brasil a prevalência média de Hb AS é próxima de 2,0% na população total, entretanto se considerarmos somente pessoas de cor negra a prevalência média é por volta de 5%, com grandes diferenças regionais. Entre pessoas de cor branca a prevalência média de Hb AS é variável entre 1,0 e 1,3%, evidenciando a representativa miscigenação branco-negra da população brasileira. Geneticamente a condição heterozigota se deve à herança do gene da globina b S por parte de um dos pais, juntamente com gene da globina b A proveniente do outro. Em nossa população as formas mais comuns de transmissão do gene b S para o traço falciforme se deve aos seguintes genótipos dos pais, por ordem decrescente de frequência: AS x AA; AS x AS; SS x AA; SC x AA; S/b Tal x AA. A deficiência de G-6PD associada à Hb AS em geral não acrescenta morbidade às já porventura preexistentes, entretanto foram relatados casos com maior incidência de hematúria e embolia pulmonar, além de discreto grau de anemia associada à presença intraeritrocitária de corpos de Heinz. A interação entre Hb AS com talassemia alfa de um ou dois genes afetados pode ser identificada pela presença de Hb H com concentrações variáveis entre 0,5 a 5%, além das hemoglobinas A2, S e A. Nesses casos é comum a diminuição da concentração da Hb S (tabela 6.18). Hematologicamente, os valores hematimétricos se situam na faixa menor dos valores de normalidade, porém é comum a presença de discreto grau de aniso-poiquilocitose no esfregaço sangüíneo. A interação de Hb AS com a doença de Hb H é uma situação extremamente rara, que apresenta a concentração da Hb H variável entre 10 e 20%, a Hb S entre 15 e 20%, e a Hb A2 visivelmente diminuída. Nesses casos a anemia é moderada a grave (Hb: 7 a 10 g/dl), com intensas alterações na morfologia eritrocitária, e precipitados de Hb H facilmente identificáveis após incubação do sangue em solução de azul crezil brilhante (figura 6.50). A tabela 6.18 resume as principais avaliações hematimétricas na interação entre Hb AS/talassemia alfa. O diagnóstico laboratorial é feito por técnicas eletroforéticas em pH alcalino (figura 6.42) e pH ácido (figura 6.43), que associadas permitem a caracterização dos genótipos de S, e em especial da Hb AS. Com o uso de gradiente de pH apropriado, a eletroforese de focalização isoelétrica também identifica a Hb S em seus diversos genótipos (figura 6.48). Outros testes que apenas detectam a presença de Hb S, sem identificar o seu genótipo (AS, SS, SC, etc.), são os testes de falcização (figura 6.51) e de solubilidade. |
||||||||||||||||||||||||||||||||||||||
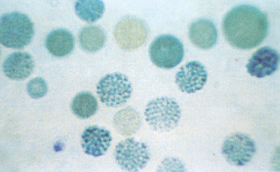
Figura 6.50 – Precipitados de Hb H na doença de Hb H, após incubação do sangue total misturado com azul de crezil brilhante a 37ºC / 1 hora. Na associação da Hb AS com doença de Hb H é possível obter resultados similares ao desta figura. |
||||||||||||||||||||||||||||||||||||||
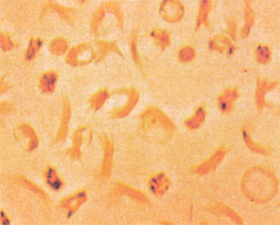
Figura 6.51 – Teste de falcização positivo em portador de traço falciforme (Hb AS) após cinco horas de incubação do sangue com metabisulfito de sódio a 1%. |
||||||||||||||||||||||||||||||||||||||
Tabela 6.18 – Relação entre genótipos de talassemia associados à Hb AS (Hb AS/tal alfa) com as alterações hematimétricas. |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
A anemia falciforme é
o estado de homozigose da Hb S representado por Hb SS. Ao contrário
dos portadores do traço falciforme (Hb AS), a história
de pacientes com anemia falciforme revela, quase invariavelmente,
por repetidos episódios dolorosos, geralmente do tipo vasculoclusivo,
com ou sem efeitos mielodepressivos (deficiência temporária
da medula óssea em produzir eritrócitos). Os sinais
e sintomas são especialmente causados por anemia crônica,
períodos de agravamento de anemia, dores nas juntas e nas
extremidades de mãos e pés, dores abdominais, necrose
asséptica da medula óssea, acidentes cerebrovasculares,
infartos pulmonares, entre outros.
Os recém-nascidos com anemia falciforme geralmente não apresentam os problemas causados por essa hemoglobinopatia devido à alta concentração de Hb Fetal presente nos eritrócitos, durante os dois primeiros meses de vida. Entretanto, a gradual substituição da Hb Fetal (a 2 g 2 ) pela Hb S (a 2 b 2S), motivada pela redução genética da síntese de globinas g ao mesmo tempo em que ocorre a indução genética da síntese de globinas b S, faz com que a doença se expresse a partir do quarto mês de vida. Os exames laboratoriais indicam anemia grave, com hemoglobina variável entre 5 e 9 g/dl, que associados à acentuada queda de hematócrito e contagem de eritrócitos resultam em índices normocíticos e normocrômicos, apesar da evidente aniso-poiquilocitose com presença de células falciformes, conforme mostram as tabelas 6.1 e 6.2. A leucocitose é moderada (12.000 a 17.000/mm3), freqüentemente com desvio à esquerda com neutrófilos bastonetes. As plaquetas podem estar discretamente elevadas. O exame da medula óssea apresenta hiperplasia normoblástica e áreas de hematopoiese podem ser encontradas em regiões geralmente ocupadas por tecido adiposo (ou medula amarela). Os exames bioquímicos do sangue revelam alterações em vários parâmetros, conforme mostra a tabela 6.19. |
||||||||||||||||||||||||||||||||||||||
Tabela 6.19 – Parâmetros bioquímicos em 74 pacientes com anemia falciforme do Instituto Estadual de Hematologia do Rio de Janeiro – HEMORIO. |
||||||||||||||||||||||||||||||||||||||
ALT – Alanina amino transferase (ou transaminase pirúvica) Fonte: Marcos Kneip Fleury, 2000. |
||||||||||||||||||||||||||||||||||||||
A eletroforese de hemoglobina apresenta concentrações de Hb S variáveis entre 90 e 100%, Hb Fetal: 2 a 10%, e Hb A2: 2 a 4%. A Hb A está sempre ausente em pacientes com anemia falciforme não transfundidos, conforme mostra a figura 6.52. |
||||||||||||||||||||||||||||||||||||||
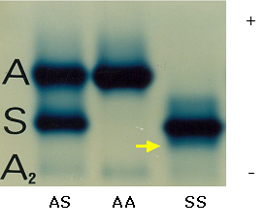
Figura 6.52 - Eletroforese alcalina de hemoglobina em acetato de celulose. (1) Hb AS; (2) Hb AA; (3) Hb SS com 5% de Hb Fetal. A seta indica fração de meta Hb S em paciente com anemia falciforme associada à deficiência de G6PD. |
||||||||||||||||||||||||||||||||||||||
A Hb Fetal na anemia falciforme – Na anemia falciforme a concentração de Hb Fetal é geralmente variável entre 2 e 10%, mas pode ser maior que 10%, e em alguns casos pode ultrapassar 40%. Pesquisas realizadas em pacientes com anemia falciforme dos sexos masculino e feminino, revelaram que o nível médio e Hb Fetal é maior entre as mulheres. A concentração de Hb Fetal é determinada por fatores relacionados e não-relacionados com o agrupamento de genes da globina beta, fato evidenciado entre os diferentes haplótipos de Hb S obtidos de estudos realizados em pessoas com anemia falciforme (ver tabela 6.3). A análise dessa relação revela que a gravidade clínica da anemia falciforme é menor quando o nível de Hb Fetal está mais elevado. A relação entre os diferentes haplótipos de Hb S com a concentração de Hb Fetal parece estar relacionada com a persistência hereditária de Hb Fetal (PHHF) dos genes específicos para síntese da globina gama. No cromossomo 11 há dois genes gama que expressam globinas gamas diferentes – um deles com a síntese de alanina e outro com a síntese de glicina – e por isso denominados por gene g A e gene g G (ver figura 6.9). Nas pequenas concentrações de Hb Fetal presente em adultos com hemoglobinas normais, a relação g G / g A, tende a um valor próximo de 2:3. Nas colônias de células eritróides obtidas de sangue fetal a Hb Fetal tem maior proporção de g G, enquanto que em colônias com maior concentração de Hb A, o predomínio é de g A. A porcentagem de células F, ou seja, eritrócitos com Hb Fetal, está aumentada na anemia falciforme. Estudos realizados com amostras de sangue de pacientes com anemia falciforme, em comparação com Hb AA, revelaram que cerca de 55% dos eritrócitos falciformes continham Hb Fetal, enquanto que em portadores de Hb AA o valor médio foi de 3%. Foi demonstrado também que fatores genéticos ligados ao cromossomo X interferem na síntese de Hb Fetal, fato que explicaria a maior concentração de Hb Fetal entre as mulheres. Hb SS / Talassemia alfa (Hb SH) – A talassemia alfa é uma alteração da síntese de globina alfa que pode ter origem genética ou adquirida. A causa hereditária da talassemia alfa está bem definida e se deve a defeitos no processo de síntese de globinas alfa, por lesão molecular de um, dois, três ou quatro genes alfa (ver figuras 4.1 e 4.2 do capítulo Hb normais). A causa adquirida tem sido relatada em pacientes com doenças linfo e mieloproliferativas, cuja prevalência de talassemia alfa chega atingir 60% entre esses doentes. Entretanto, pouco se sabe de que forma surge a talassemia alfa adquirida – se induzida por drogas quimioterápicas ou por alterações genéticas da célula tronco hematopoiética. Entre pessoas negras, a talassemia alfa resulta geralmente da deleção de um ou dois genes alfa. A prevalência de talassemia alfa entre negros da América do Norte é de 30%, enquanto no Brasil é variável entre 20 e 25%. Assim, admite-se que a interação entre a anemia falciforme e talassemia alfa ocorra com alta frequência. Foi demonstrado em vários estudos descritos na literatura científica que a talassemia alfa representa um fator modulador dos parâmetros hematológicos, com elevação da contagem de eritrócitos e redução dos índices hematimétricos: VCM (volume corpuscular médio), HCM (hemoglobina corpuscular média), e CHCM (concentração de hemoglobina corpuscular média). A redução do número de moléculas de Hb S no eritrócito diminui a formação de polímeros das moléculas de desoxi-HbS, e consequentemente se forma menos eritrócitos falciformes irreversíveis. Dessa forma, a interação entre Hb S/talassemia alfa parece ser benéfica à anemia falciforme pois aumenta a sobrevida do eritrócito falcêmico, diminui o grau de hemólise, e consequentemente torna a anemia menos grave, conforme mostra a tabela 6.20. Entretanto, na associação entre Hb S e doença de Hb H (lesão de três genes alfa) a anemia se torna grave. Nesses casos a concentração de Hb H associada à Hb S é variável entre 15 e 25% (figura 6.53). |
||||||||||||||||||||||||||||||||||||||
Tabela 6.20 – Parâmetros hematológicos na interação entre anemia falciforme e talassemia alfa. |
||||||||||||||||||||||||||||||||||||||
** Lesão de dois genes alfa; Hb H: 2 a 8% na eletroforese alcalina. *** Lesão de três genes alfa ou Hb S/doença de Hb H; Hb H: 15 a 25% na eletroforese alcalina. |
||||||||||||||||||||||||||||||||||||||
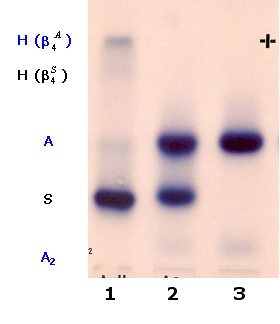
Figura 6.53 - Eletroforese alcalina de hemoglobina em acetato de celulose. (1): Hb SH obtida de paciente com anemia falciforme (transfundida, por isso a presença de Hb A) associada a talassemia alfa (- a b 2S / - - b 2S), observe a presença de duas frações de Hb H: uma formada por tetrâmeros de b A e outra por tetrâmeros de b S; a concentração maior da Hb H por b 4A em relação à diminuta concentração da Hb H por b 4S se deve ao fato deste tetrâmero ser muito instável. (2): Hb AS. (3): Hb AA. Para visualizar a Hb H é necessário aplicar hemolisado feito com saponina a 1%. |
||||||||||||||||||||||||||||||||||||||
Anemia falciforme e deficiência de G-6-PD – A coexistência da Hb SS com deficiência de glicose-6-fosfato desidrogenase (G-6-PD) é um fato presumível uma vez que a prevalência da deficiência dessa enzima entre a população negra é por volta de 9 a 15%. Embora a literatura científica se refere de forma genérica que a associação entre Hb SS/deficiência de G-6-PD parece não ter influência no número de eritrócitos falciformes irreversíveis (ou células densa), ou no nível de concentração da hemoglobina total, bem como na condição clínica do paciente, é importante destacar que há, pelo menos, cinco classes diferentes de deficiência de G-6-PD (tabela 6.21). A classe 1 é muito rara, entretanto as classes 2 e 3 são as mais importantes sob o ponto de vista clínico, pois causam crises de hemólises quando o portador é exposto a drogas oxidantes. No Brasil, a deficiência de G-6-PD mais comum é a da classe 3, com os tipos africanos prevalecendo em 87% dos casos descritos, seguida da classe 2 onde os tipos mediterrâneos atingem 13%. |
||||||||||||||||||||||||||||||||||||||
Tabela 6.21 – Classificação da deficiência de G-6-PD em cinco classes. |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
Sob
o ponto de vista fisiológico a associação da
Hb SS com deficiência de G-6-PD é desastrosa à
molécula de hemoglobina. A deficiência de G-6-PD é
um fator indutor da desnaturação da hemoglobina, enquanto
que a Hb S apresenta alto grau de oxidação expontânea.
A associação desses dois defeitos num mesmo eritrócito
certamente desencadeia com maior intensidade a desnaturação
oxidativa da Hb S, especialmente se exposto a fatores indutores
como são os casos de drogas e poluentes ambientais oxidativos.
Diante desses fatos ocorre maior consumo das enzimas antioxidantes,
alterando as sínteses e as suas concentrações.
Assim, os mecanismos redutores da oxidação e das lesões
celulares provocadas pelos radicais livres causam a diminuição
das atividades de glutatião peroxidase e catalase, aumenta
o nível de superóxido dismutase (SOD) e diminui a
concentração de vitamina E no plasma e na membrana
eritrocitária. A avaliação citológica de corpos de Heinz nos pacientes com anemia falciforme é sempre um excelente marcador biológico dos processos oxidativos. Na associação entre Hb SS e deficiência de G-6-PD a precipitação intraeritrocitária de corpos de Heinz é muito mais expressiva. Outro indicador da deficiência de G-6-PD em pacientes com anemia falciforme é a elevada concentração de metaemoglobina, que pode ser quantificada bioquimicamente ou visualizada durante o processo eletroforético da Hb SS – notadamente após 48 horas da coleta do sangue (figura 6.52). Investigação laboratorial – O diagnóstico laboratorial da anemia falciforme inclui o hemograma completo, as eletroforeses de hemoglobina em tampões alcalino e ácido, dosagens de Hb Fetal e metaemoglobina, contagem de reticulócitos e pesquisas intraeritrocitárias de Hb H e corpos de Heinz. O hemograma é fundamental para avaliar quantitativamente o grau de anemia, além de fornecer os índices hematimétricos de VCM e HCM que são importantes parâmetros para diferenciar os genótipos da anemia falciforme (SS e SF) e da talassemia beta (SF), conforme mostram as tabelas 6.1e 6.17. A eletroforese de hemoglobina em pH alcalino identifica a fração anormal com suspeita de ser a Hb S, pois nesta mesma região eletroforética se posiciona a Hb D. Assim, os genótipos SS, SD e DD apresentam com as mesmas características eletroforéticas, conforme mostra a figura 5.5 do capítulo 5 - Hemoglobinopatias. Para identificar cada um desses genótipos é necessário recorrer à eletroforese de hemoglobinas em gel de agarose com pH ácido, que separa com excelente resolução esses genótipos. É importante destacar ainda que a eletroforese alcalina, quer seja em gel de agarose ou em acetato de celulose, permite a separação da Hb Fetal e sua quantificação densitométrica, fato que auxilia o diagnóstico dos diferentes genótipos das doenças falciformes. Por outro lado, a eletroforese alcalina em acetato de celulose é fundamental na identificação da Hb H e da metaemoglobina (que apresenta cor marrom) quando associadas à anemia falciforme. A dosagem de Hb Fetal é importante não apenas para caracterizar o genótipo da doença falciforme, mas principalmente para o entendimento da evolução clínica do paciente, onde o maior nível de Hb Fetal representa melhor prognóstico da doença. Por outro lado, a dosagem da metaemoglobina indica o grau de oxidação da Hb S, pois as concentrações elevadas estão relacionadas com processos fisiopatológicos mais deletérios ao eritrócito falciforme, bem como podem indicar a associação da Hb SS com deficiência de G-6-PD; nestes casos se faz necessário a dosagem enzimática desta enzima. A contagem de reticulócitos estabelece importantes parâmetros sobre o processo hemolítico decorrente da anemia falciforme, bem como a resposta da reposição eritrocitária – ou eritropoiese. A pesquisa intraeritrocitária de Hb H (figura 6.50) é um excelente método de confirmação da talassemia alfa. A pesquisa intraeritrocitária de corpos de Heinz representa importante informação sobre o desencadeamento de processos oxidativos da Hb S, bem como a suspeita da associação entre Hb SS e deficiência de G-6-PD. Finalmente, é sempre importante enfatizar que para a correta conclusão do diagnóstico laboratorial da Hb SS ou da Hb SS/Talassemia alfa é necessário que se faça a análise de hemoglobinas no sangue dos pais. No caso da Hb SS, os genótipos mais comuns dos pais são: AS x AS; AS x SS; AS x SC; AS x S/Talassemia Beta. Quando a Hb SS está associada à talassemia alfa um dos pais deve apresentar a Hb H, por exemplo: AS x ASH; ASH x SC, etc. Interação Hb S/Talassemia beta
|
||||||||||||||||||||||||||||||||||||||
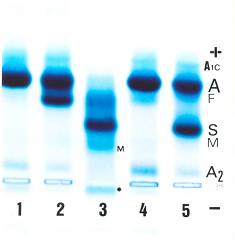
Figura 6.54 - Eletroforese alcalina de hemoglobinas em gel de agarose. (1) Hb AA – padrão normal; (2) Hb AF na talassemia beta maior após transfusão de concentrado de hemácias; (3) Hb S/Tal. b 0 ou Hb SF com excesso de metaemoglobina S e globinas alfa livres atrás do local da aplicação; (4) Pai do caso 3 – talassemia beta menor com Hb A2 e Hb Fetal aumentadas; (5) Mãe do caso 3 com Hb AS. |
||||||||||||||||||||||||||||||||||||||
Esse genótipo se deve à total deficiência de síntese de globina beta normal ou b A, sendo por isso denominado por talassemia b 0 que associado a Hb S se torna Hb S /Tal.b 0. Na talassemia b 0 há elevada expressão do gene gama que induz o aumento da síntese da Hb Fetal quando há interação com a Hb S. A situação mais freqüente de herança desse genótipo ocorre quando um dos pais é portador heterozigoto de talassemia beta menor, ou Tal. b 0 heterozigoto, e o outro é portador de Hb AS (figura 6.55). Pela representação esquemática dessa figura é possível entender a ausência eletroforética da Hb A, e as concentrações elevadas das Hb S e Hb Fetal. Entretanto um fato relevante mostrado no esquema se refere às globinas alfa "livres" que por não terem globinas b A para se combinarem, precipitam nos eritrócitos e formam corpos de Heinz. Os corpos de Heinz são retirados por macrófagos do sistema retículo endotelial, causando lesões nos eritrócitos – principal causa da formação de micrócitos esquisócitos e, consequentemente da microcitose. As globinas alfa "livres" são facilmente identificadas em eletroforese alcalina de hemoglobina em agarose ou acetato de celulose, que após coloração com negro de amido são visíveis atrás do ponto de aplicação das amostras (figura 6.54). Destaque importante dessa figura se deve à presença de meta Hb S induzida pelo alto grau de oxidação da Hb S e também, em parte causada pela oxidação das globinas alfa "livres". Como já foi exposto anteriormente, o alto grau de oxidação da hemoglobina inviabiliza a sua integridade molecular, tornando-a instável estruturalmente – principal causa da precipitação de globinas bS sob forma de corpos de Heinz. |
||||||||||||||||||||||||||||||||||||||
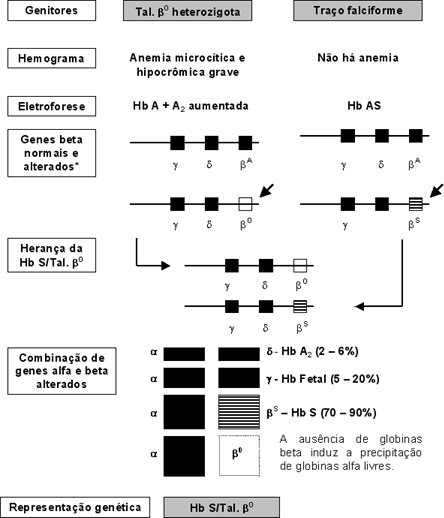
* - Nesta representação só foram incluídos os genes do cromossomo 11 (g, d e b). Os quatro genes alfa não foram incluídos, admitindo-os com sínteses normais. Figura 6.55 – Representação esquemática da herança e das conseqüências fisiopatológicas da Hb S/Tal. b 0. |
||||||||||||||||||||||||||||||||||||||
O genótipo SFA se deve à associação entre a talassemia b + - caso em que o gene da globina b A está parcialmente afetado para sintetizar Hb A – e Hb S, configurando a representação genética de Hb S/Tal.b +. Nesse caso a forma mais comum da expressão dessas alterações se caracteriza pela maior concentração da Hb S em relação à Hb A (figura 6.56). Eventualmente a Hb Fetal pode estar elevada. Pelo fato de ocorrer alguma síntese de Hb A, que pode representar entre 10 e 40% do total da hemoglobina (ver tabela 6.1), as conseqüências fisiopatológicas são menores. |
||||||||||||||||||||||||||||||||||||||
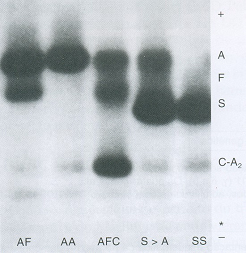
Figura 6.56 – Eletroforese alcalina de hemoglobinas em acetato de celulose. Da esquerda para a direita: AF (talassemia beta maior após transfusão de concentrado de hemácias); AA (padrão normal); AFC (recém-nascido com Hb AC e Hb Fetal elevada devido à idade); S>A (Hb S/Tal. b +, com Hb S mais concentrada que a Hb A; SS (Anemia falciforme). |
||||||||||||||||||||||||||||||||||||||
A figura 6.57 apresenta o esquema da herança e conseqüências fisiopatológicas da Hb S/Tal. b +. Devido a presença de Hb A – mesmo que em baixas concentrações – o nível de globinas alfa livres é nitidamente inferior em comparação com o caso anterior. |
||||||||||||||||||||||||||||||||||||||
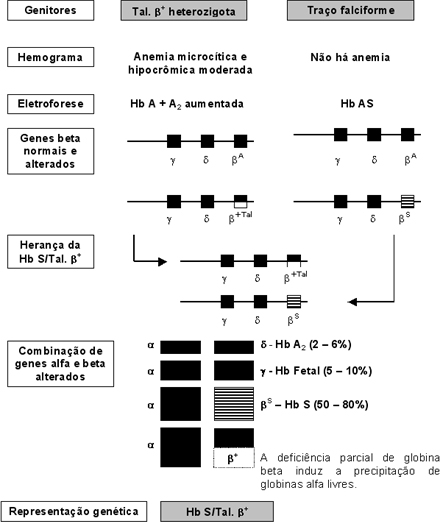
* - Nesta representação só foram incluídos os genes do cromossomo 11 (g, d e b). Os quatro genes alfa não foram incluídos, admitindo-os com sínteses normais. Figura 6.57 – Representação esquemática da herança e das conseqüências fisiopatológicas da Hb S/Tal. b +. |
||||||||||||||||||||||||||||||||||||||
É fundamental a realização dos exames eletroforéticos dos pais de portadores de Hb S/Tal. b, onde um deles deve apresentar Hb S (Hb AS, Hb SC, Hb SS ou Hb S/Tal. b) e o outro talassemia beta (Tal. b menor, Hb S/Tal. b ou Hb C/Tal. b). Entretanto os genótipos mais comuns dos pais são: Hb AS x Hb Ab (Tal. b menor). Essa avaliação é necessária porque muitas vezes é transfundido sangue com Hb AA para pessoas com anemia falciforme (Hb SS), e nesses casos o resultado eletroforético pós-transfusional é similar ao da Hb S/Tal. b+, com a concentração da Hb S maior que a Hb A. Além do hemograma, com destaque para os índices VCM, HCM e resultados das análises por eletroforeses alcalina e ácida, é necessário que se faça a dosagem de Hb Fetal para o correto estabelecimento do diagnóstico laboratorial da Hb S/Talassemia Beta. A dosagem de metaemoglobina, contagem de reticulócitos, e pesquisa intraeritrocitária de corpos de Heinz constituem excelentes parâmetros para avaliar as conseqüências fisiopatológicas da doença. Hb SD – Doença da Hb SD A Hb D é uma hemoglobina
variante com a mesma mobilidade eletroforética da Hb S (figura
5.5 do capítulo 5 - Hemoglobinopatias). Há vários
tipos estruturalmente diferentes de Hb D, conforme mostra a tabela
5.3 do capítulo 5 - Hemoglobinopatias. Desses tipos a Hb
D Los Angeles, também conhecida por D Punjab, é mais
comum em várias regiões do mundo, inclusive no Brasil,
onde sua prevalência no estado de heterozigose (Hb AD) ocorre
na proporção de um caso para cada 5 mil pessoas analisadas.
Por essa razão não é difícil encontrar
pessoas com a dupla heterozigose entre hemoglobinas S e D (Hb SD)
padecendo de anemia hemolítica crônica, e por isso
identificadas como doença de Hb SD. A anemia é variável
entre moderada e grave, uma vez que a Hb D participa com certa intensidade
do processo de polimerização desencadeado pela desoxi-Hb
S. As tabelas 6.1 e 6.2, mostram características laboratoriais
da Hb SD. Pelo fato da Hb SD ter e mesma mobilidade eletroforética
das Hb SS e Hb DD nas eletroforeses alcalina de hemoglobinas em
gel de agarose e acetato de celulose, a melhor forma de diferencia-las
é por meio da eletroforese ácida em gel de agarose
(figura 5.5, capítulo 5). As concentrações
das frações de Hb S e Hb D são equilibradas,
oscilando cada uma delas entre 45 e 50%. Além do hemograma
e das eletroforeses alcalina e ácida, a contagem de reticulócitos
é fundamental para avaliar o grau de eritropoiese do paciente
com Hb SD. O gene da globina b C é comum na costa oeste da África, com alta prevalência em Benin, Togo e Gana – regiões que no período do tráfico de escravos constituíam as Costas do Ouro e do Marfim, e de onde vieram grande número de africanos para o Brasil. Assim, a prevalência da Hb AC na população brasileira oscila entre 0,5 e 1%, e entre pessoas de cor negra entre 1 e 3%. Há dois tipos de Hb C descritas na literatura, a Hb C com mutação no aminoácido n.º 6 da globina beta caracterizada pela troca de ácido glutâmico por lisina (b 6 Glu ® Lis), e a Hb C Harlem com dupla mutação (b 6 Glu ® Lis + b 73 Asp ® Asn). A Hb C Harlem é muito rara em todo o mundo, e também no Brasil, e por ter dupla mutação, uma das quais similar à da Hb S, pode causar discreta falcização dos eritrócitos. Dessa forma, a Hb C (b 6 Glu ® Lis) é a que tem sido descrita na dupla heterozigose com a Hb S, causando a doença de Hb SC, com anemia hemolítica crônica com graus variáveis entre moderado a discreto. As alterações laboratoriais podem ser avaliadas nas tabelas 6.1 e 6.2, com destaques para o elevado número de células em alvo e presença de cristais de Hb C nos eritrócitos. Devido à clínica da Hb SC, gestantes portadoras dessa dupla heterozigose devem ter acompanhamento especial durante o período de gestação. A análise laboratorial específica é realizada por meio de eletroforese alcalina de hemoglobinas em gel de agarose ou em acetato de celulose, com a Hb C separando-se da Hb S e se posicionando junto com a Hb A2 (figura 6.42). Em eletroforese ácida em gel de agarose a Hb C também é mais lenta que a Hb S (figura 6.58). Pelo fato da mobilidade da Hb C em eletroforese alcalina ser similar às da Hb O Arábia (comum no Oriente Médio) e Hb E (comum no Sudeste Asiático), é importante o uso da eletroforese ácida em gel de agarose, uma vez que nessas condições a Hb O Arábia e Hb E apresentam mobilidade igual à da Hb A, diferenciando-se, portanto, da Hb C. |
||||||||||||||||||||||||||||||||||||||
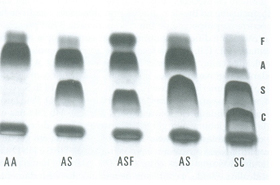
Figura 6.58 – Eletroforese ácida de hemoglobinas em gel de agarose. (1) Hb AA; (2) Hb AS; (3) Hb ASF em sangue de recém-nascido; (4) Hb AS; (5) Hb SC com traços de Hb A proveniente de transfusão de concentrado de hemácias há mais de dois meses. |
||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||